WSGR Docket No.55776-726.601 MIXED SEROTONIN RECEPTOR BINDERS FOR TREATMENT OF PSYCHOTIC DISORDERS CROSS-REFERENCE TO RELATED APPLICATIONS [0001] This application claims the benefit of U.S. Provisional Application No.63/614,457, filed December 22, 2023, and U.S. Provisional Application No.63/735,155, filed December 17, 2024, each of which is incorporated herein by reference in its entirety. FIELD OF THE INVENTION [0002] Described herein are compounds, methods of making such compounds, pharmaceutical compositions and medicaments comprising such compounds, and methods of using such compounds for the treatment of conditions, diseases, or disorders. BACKGROUND [0003] Over the last several decades, predominant interest in serotonergic targets with respect to schizophrenia has centered around 5-HT2A antagonism/inverse agonism.5-HT2C receptor agonists have emerged as additional serotonergic target of interest.5-HT2C agonists have been suggested as treatments for multiple symptom domains of schizophrenia including positive, negative, cognitive, and depressive symptoms without the adverse events or tolerability issues associated with existing agents. The 5-HT2C receptor is a highly complex, highly regulated receptor which is widely distributed throughout the brain. The 5-HT2C receptor couples to multiple signal transduction pathways leading to engagement of a number of intracellular signaling molecules. Moreover, there are multiple allelic variants of the 5-HT2C receptor and the receptor is subject to RNA editing in the coding regions. The complexity of this receptor is further emphasized by the utility of either agonists or antagonists in the treatment of schizophrenia. The preclinical profile of 5-HT2C agonists from a neurochemical, electrophysiological, and a behavioral perspective is indicative of antipsychotic-like efficacy. Selective 5-HT2C agonist vabicaserin demonstrated clinical efficacy in a Phase II trial in schizophrenia patients. These data suggest that 5-HT2C agonists are potential therapeutics for the treatment of psychiatric disorders, including psychotic disorders. SUMMARY [0004] In one aspect, disclosed herein are compounds having dual 5-HT2a antagonist and 5-HT2c agonist activity. In one aspect, disclosed herein is a compound of Formula (I), or a pharmaceutically acceptable salt, pharmaceutically acceptable solvate, diastereomeric mixture, individual enantiomer, or isotopically enriched form thereof:
WSGR Docket No.55776-726.601

Formula (I) wherein: each R
3, R
4, and R
5 is independently hydrogen, C
1-C
6alkyl, C
1-C
6haloalkyl, C
1-C
6hydroxyalkyl, C
1- C
6aminoalkyl, C
1-C
6heteroalkyl, halogen, -CN, -NO
2, -OR
a, -SR
a, -NR
cR
d, -S(=O)R
b, -S(=O)
2R
b, - S(=O)
2NR
cR
d, -NR
bS(=O)
2R
b, -NR
bS(=O)
2NR
cR
d, -C(=O)R
b, -C(=O)OR
b, -OC(=O)R
b, - OC(=O)OR
b, -OC(=O)NR
cR
d, -NR
bC(=O)R
b, -NR
bC(=O)OR
b, -NR
bC(=O)NR
cR
d, -C(=O)NR
cR
d, - P(=O)(OR
c)(OR
d), -P(=O)R
cR
d, aryl, heteroaryl, cycloalkyl, heterocycloalkyl, C
1-C
6alkyl(cycloalkyl), C
1-C
6alkyl(heterocycloalkyl), C
1-C
6alkyl(aryl), or C
1-C
6alkyl(heteroaryl), wherein each of the alkyl, heteroalkyl, aryl, heteroaryl, cycloalkyl, and heterocycloalkyl is optionally substituted with one or more substituents selected from C
1-C
3alkyl, C
1-C
3haloalkyl, C
1- C
3alkoxy, halogen, -OH, -CN, and =O; R
8 is hydrogen or C
1-C
6alkyl; R
9 is hydrogen or C
1-C
6alkyl; or R
8 and R
9 are taken together to form a heterocycloalkyl, wherein the heterocycloalkyl is optionally substituted with one or more substituents selected from C1-C3alkyl, C1-C3haloalkyl, C1-C3alkoxy, halogen, -OH, and =O; each R
a is independently hydrogen, C1-C6alkyl, C1-C6haloalkyl, C1-C6heteroalkyl, aryl, C1- C3alkyl(phenyl), C3-C6cycloalkyl, 5- to 6- membered heteroaryl, C1-C3alkyl(5- to 6- membered heteroaryl), or 4- to 6- membered heterocycloalkyl, wherein each alkyl, heteroalkyl, aryl, cycloalkyl, heteroaryl, and heterocycloalkyl is optionally substituted with one or more substituents selected from C1-C3alkyl, C1-C3haloalkyl, C1-C3alkoxy, halogen, -OH, -CN, and =O; each R
b is independently hydrogen or C1-C6alkyl; abd each R
c and R
d is independently hydrogen or C1-C6alkyl; or R
c and R
d are taken together to form a 4- to 6- membered heterocycloalkyl, wherein the heterocycloalkyl is optionally substituted with one or more substituents selected from C1-C3alkyl, C1-C3haloalkyl, C1- C3alkoxy, halogen, -OH, -CN, and =O; wherein at least two of R
3, R
4, and R
5 are not hydrogen.
WSGR Docket No.55776-726.601 [0005] In some embodiments, each R
3, R
4, and R
5 is independently hydrogen, C
1-C
6alkyl, C
1-C
6haloalkyl, C
1-C
6hydroxyalkyl, C
1-C
6aminoalkyl, C
1-C
6heteroalkyl, halogen, -CN, -NO
2, -OR
a, -SR
a, -NR
cR
d, -S(=O)R
b, -S(=O)
2R
b, -S(=O)
2NR
cR
d, -NR
bS(=O)
2R
b, -NR
bS(=O)
2NR
cR
d, -C(=O)R
b, -C(=O)OR
b, -OC(=O)R
b, - OC(=O)OR
b, -OC(=O)NR
cR
d, -NR
bC(=O)R
b, -NR
bC(=O)OR
b, -NR
bC(=O)NR
cR
d, -C(=O)NR
cR
d, - P(=O)(OR
c)(OR
d), -P(=O)R
cR
d, C
6-C
10aryl, 5-10 membered heteroaryl, C
3-C
7cycloalkyl, 3- to 10- membered heterocycloalkyl, C
1-C
6alkyl(C
3-C
7cycloalkyl), C
1-C
6alkyl(3- to 10- membered heterocycloalkyl), C
1-C
6alkyl(C
6-C
10aryl), or C
1-C
6alkyl(heteroaryl), wherein each of the alkyl, heteroalkyl, aryl, heteroaryl, cycloalkyl, and heterocycloalkyl is optionally substituted with one or more substituents selected from C
1- C3alkyl, C1-C3haloalkyl, C1-C3alkoxy, halogen, -OH, -CN, and =O. [0006] In some embodiments, provided herein is the compound of Formula (I), or a pharmaceutically acceptable salt, pharmaceutically acceptable solvate, diastereomeric mixture, individual enantiomer, or isotopically enriched form thereof, for use as medicine. [0007] In one aspect, disclosed herein is a pharmaceutical composition comprising the compound of Formula (I), or a pharmaceutically acceptable salt, pharmaceutically acceptable solvate, diastereomeric mixture, individual enantiomer, or isotopically enriched form thereof. In some embodiments, the pharmaceutical composition further comprises at least one pharmaceutically acceptable excipient. [0008] In one aspect, disclosed herein is a method of treating a disease or disorder in a subject in need thereof, wherein the method comprises administering to the subject a therapeutically effective amount of a compound disclosed herein (e.g., a compound of Formula (I)), or a pharmaceutically acceptable salt, pharmaceutically acceptable solvate, diastereomeric mixture, individual enantiomer, or isotopically enriched form thereof. In some embodiments, the disease or disorder is a disease or disorder of the brain. In some embodiments, the neurological disease or disorder is a neurodegenerative, a neuropsychiatric, or substance use disease or disorder. In some embodiments, the neurological disease or disorder is an injury. In some embodiments, the disease or disorder is an anxiety disorder, a mood disorder, a psychotic disorder, a personality disorder, an eating disorder, a sleep disorder, a sexuality disorder, an impulse control disorder, a substance use disorder, a dissociative disorder, a cognitive disorder, a developmental disorder, or a factitious disorder. In some embodiments, the disease or disorder is a psychotic disorder. In some embodiments, the psychotic disorder is selected from schizophrenia, schizoaffective disorder, schizophreniform disorder, brief psychotic disorder, delusional disorder, shared psychotic disorder, substance-induced psychotic disorder, paraphrenia, psychotic depression, bipolar disorder, schizotypal personality disorder, paranoid personality disorder, schizoid personality disorder, borderline personality disorder, post-traumatic stress disorder, obsessive-compulsive disorder, and dissociative disorders, or psychosis associated with a neurodegenerative disorders. In some embodiments, the neurodegenerative disorder is selected from Huntington’s disease, Alzheimer’s disease, Lewy body dementia, and Parkinson’s disease In some embodiments, the psychotic disorder is schizophrenia or bipolar disorder. In some embodiments, the method further comprises administering to the subject a therapeutically effective amount of an additional therapeutic agent.
WSGR Docket No.55776-726.601 [0009] Other objects, features and advantages of the compounds, methods and compositions described herein will become apparent from the following detailed description. It should be understood, however, that the detailed description and the specific examples, while indicating specific embodiments, are given by way of illustration only, since various changes and modifications within the spirit and scope of the instant disclosure will become apparent to those skilled in the art from this detailed description. DETAILED DESCRIPTION [0010] The present disclosure provides non-hallucinogenic compounds useful for the treatment of a variety of brain disorders, including psychotic disorders, as well as for increasing neuronal plasticity. [0011] Compounds capable of modifying neural circuits have potential for treating neurological diseases and disorders that are mediated by the loss of synaptic connectivity and/or plasticity. Moreover, such compounds are likely to produce sustained therapeutic effects because, for example, of the potential to treat the underlying pathological changes in circuitry. Certain Terminology [0012] Unless otherwise stated, the following terms used in this application have the definitions given below. It must be noted that, as used in the specification and the appended claims, the singular forms “a”, “an”, and “the” include plural referents unless the context clearly dictates otherwise. In this application, the use of “or” means “and/or” unless stated otherwise. The use of the term “including” as well as other forms, such as “include,” “includes,” and “included,” is not limiting. The section headings used herein are for organizational purposes only and are not to be construed as limiting the subject matter described. [0013] As used herein, C1-Cx (or C1-x) includes C1-C2, C1-C3... C1-Cx. By way of example only, a group designated as "C1-C4" indicates that there are one to four carbon atoms in the moiety, i.e., groups containing 1 carbon atom, 2 carbon atoms, 3 carbon atoms or 4 carbon atoms. Thus, by way of example only, "C1-C4 alkyl" indicates that there are one to four carbon atoms in the alkyl group, i.e., the alkyl group is selected from among methyl, ethyl, propyl, iso-propyl, n-butyl, iso-butyl, sec-butyl, and t-butyl. [0014] “Alkyl” generally refers to a straight or branched hydrocarbon chain radical consisting solely of carbon and hydrogen atoms, such as having from one to fifteen carbon atoms (e.g., C1-C15 alkyl). Unless otherwise state, alkyl is saturated or unsaturated (e.g., an alkenyl, which comprises at least one carbon- carbon double bond). Disclosures provided herein of an “alkyl” are intended to include independent recitations of a saturated “alkyl,” unless otherwise stated. Alkyl groups described herein are generally monovalent but may also be divalent (which may also be described herein as “alkylene” or “alkylenyl” groups). In certain embodiments, an alkyl comprises one to thirteen carbon atoms (e.g., C
1-C
13 alkyl). In certain embodiments, an alkyl comprises one to eight carbon atoms (e.g., C1-C8 alkyl). In other embodiments, an alkyl comprises one to five carbon atoms (e.g., C
1-C
5 alkyl). In other embodiments, an alkyl comprises one to four carbon atoms (e.g., C
1-C
4 alkyl). In other embodiments, an alkyl comprises one to three carbon atoms (e.g., C
1-C
3 alkyl). In other embodiments, an alkyl comprises one to two carbon atoms (e.g., C
1-C
2 alkyl). In other embodiments, an alkyl comprises one carbon atom (e.g., C
1 alkyl). In other
WSGR Docket No.55776-726.601 embodiments, an alkyl comprises five to fifteen carbon atoms (e.g., C
5-C
15 alkyl). In other embodiments, an alkyl comprises five to eight carbon atoms (e.g., C
5-C
8 alkyl). In other embodiments, an alkyl comprises two to five carbon atoms (e.g., C
2-C
5 alkyl). In other embodiments, an alkyl comprises three to five carbon atoms (e.g., C
3-C
5 alkyl). In other embodiments, the alkyl group is selected from methyl, ethyl, 1-propyl (n- propyl), 1-methylethyl (iso-propyl), 1-butyl (n-butyl), 1-methylpropyl (sec-butyl), 2-methylpropyl (iso- butyl), 1,1-dimethylethyl (tert-butyl), 1-pentyl (n-pentyl). The alkyl is attached to the rest of the molecule by a single bond. In general, alkyl groups are each independently substituted or unsubstituted. Each recitation of “alkyl” provided herein, unless otherwise stated, includes a specific and explicit recitation of an unsaturated “alkyl” group. Similarly, unless stated otherwise specifically in the specification, an alkyl group is optionally substituted by one or more of the following substituents: halo, cyano, nitro, oxo, thioxo, imino, oximo, trimethylsilanyl, -OR
x, -SR
x, -OC(O)-R
x, -N(R
x)2, -C(O)R
x, -C(O)OR
x, -C(O)N(R
x)2, - N(R
x)C(O)OR
x, -OC(O)-N(R
x)2, -N(R
x)C(O)R
x, -N(R
x)S(O)tR
x (where t is 1 or 2), -S(O)tOR
x (where t is 1 or 2), -S(O)tR
x (where t is 1 or 2) and -S(O)tN(R
x)2 (where t is 1 or 2) where each R
x is independently hydrogen, alkyl (optionally substituted with halogen, hydroxy, methoxy, or trifluoromethyl), fluoroalkyl, carbocyclyl (optionally substituted with halogen, hydroxy, methoxy, or trifluoromethyl), carbocyclylalkyl (optionally substituted with halogen, hydroxy, methoxy, or trifluoromethyl), aryl (optionally substituted with halogen, hydroxy, methoxy, or trifluoromethyl), aralkyl (optionally substituted with halogen, hydroxy, methoxy, or trifluoromethyl), heterocyclyl (optionally substituted with halogen, hydroxy, methoxy, or trifluoromethyl), heterocyclylalkyl (optionally substituted with halogen, hydroxy, methoxy, or trifluoromethyl), heteroaryl (optionally substituted with halogen, hydroxy, methoxy, or trifluoromethyl), or heteroarylalkyl (optionally substituted with halogen, hydroxy, methoxy, or trifluoromethyl). In some embodiments, an alkyl group is substituted with one or more fluorine. [0015] An “alkylene” group refers to a divalent alkyl radical. Any of the above-mentioned monovalent alkyl groups may be an alkylene by abstraction of a second hydrogen atom from the alkyl. In some embodiments, an alkylene is a C1-C6alkylene. In other embodiments, an alkylene is a C1-C4alkylene. Typical alkylene groups include, but are not limited to, -CH2-, -CH(CH3)-, -C(CH3)2-, -CH2CH2-, -CH2CH(CH3)-, - CH2C(CH3)2-, -CH2CH2CH2-, -CH2CH2CH2CH2-, and the like. Unless stated otherwise specifically in the specification, an alkylene chain is optionally substituted as described for alkyl groups herein. [0016] The term “alkenyl” refers to a type of alkyl group in which at least one carbon-carbon double bond is present. In one embodiment, an alkenyl group has the formula –C(R)=CR2, wherein R refers to the remaining portions of the alkenyl group, which may be the same or different. In some embodiments, R is H or an alkyl. Non-limiting examples of an alkenyl group include -CH=CH
2, -C(CH
3)=CH
2, -CH=CHCH
3, - C(CH3)=CHCH3, and –CH2CH=CH2. [0017] The term “alkynyl” refers to a type of alkyl group in which at least one carbon-carbon triple bond is present. In one embodiment, an alkenyl group has the formula -C≡C-R, wherein R refers to the remaining portions of the alkynyl group. In some embodiments, R is H or an alkyl. Non-limiting examples of an alkynyl group include -C≡CH, -C≡CCH
3 -C≡CCH
2CH
3, -CH
2C≡CH.
WSGR Docket No.55776-726.601 [0018] An “alkoxy” group refers to a (alkyl)O- group, where alkyl is as defined herein. [0019] The term “alkylamine” refers to -NH(alkyl), or -N(alkyl)
2. [0020] The term “aromatic” refers to a planar ring having a delocalized ^-electron system containing 4n+2 ^ electrons, where n is an integer. The term “aromatic” includes both carbocyclic aryl (“aryl,” e.g., phenyl) and heterocyclic aryl (or “heteroaryl” or “heteroaromatic”) groups (e.g., pyridine). The term includes monocyclic or fused-ring polycyclic (i.e., rings which share adjacent pairs of carbon atoms) groups. [0021] The term “carbocyclic” or “carbocycle” refers to a ring or ring system where the atoms forming the backbone of the ring are all carbon atoms. The term thus distinguishes carbocyclic from “heterocyclic” rings or “heterocycles” in which the ring backbone contains at least one atom which is different from carbon. In some embodiments, at least one of the two rings of a bicyclic carbocycle is aromatic. In some embodiments, both rings of a bicyclic carbocycle are aromatic. In certain embodiments, a carbocyclyl comprises three to ten carbon atoms. In other embodiments, a carbocyclyl comprises five to seven carbon atoms. The carbocyclyl is attached to the rest of the molecule by a single bond. Carbocyclyl or cycloalkyl is saturated (i.e., containing single C-C bonds only) or unsaturated (i.e., containing one or more double bonds or triple bonds). Examples of saturated cycloalkyls include, e.g., cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, and cyclooctyl. An unsaturated carbocyclyl is also referred to as "cycloalkenyl." Examples of monocyclic cycloalkenyls include, e.g., cyclopentenyl, cyclohexenyl, cycloheptenyl, and cyclooctenyl. Polycyclic carbocyclyl radicals include, for example, adamantyl, norbornyl (i.e., bicyclo[2.2.1]heptanyl), norbornenyl, decalinyl, 7,7-dimethyl-bicyclo[2.2.1]heptanyl, and the like. Unless otherwise stated specifically in the specification, the term "carbocyclyl" is meant to include carbocyclyl radicals that are optionally substituted by one or more substituents independently selected from alkyl, alkenyl, alkynyl, halo, fluoroalkyl, oxo, thioxo, cyano, nitro, optionally substituted aryl, optionally substituted aralkyl, optionally substituted aralkenyl, optionally substituted aralkynyl, optionally substituted carbocyclyl, optionally substituted carbocyclylalkyl, optionally substituted heterocyclyl, optionally substituted heterocyclylalkyl, optionally substituted heteroaryl, optionally substituted heteroarylalkyl, -R
y-OR
x, -R
y-OC(O)-R
x, -R
y- OC(O)-OR
x, -R
y-OC(O)-N(R
x)2, -R
y-N(R
x)2, -R
y-C(O)R
x, -R
y-C(O)OR
x, -R
y-C(O)N(R
x)2, -R
y-O-R
z- C(O)N(R
x)
2, -R
y-N(R
x)C(O)OR
x, -R
y-N(R
x)C(O)R
x, -R
y-N(R
x)S(O)
tR
x (where t is 1 or 2), -R
y-S(O)
tR
x (where t is 1 or 2), -R
y-S(O)
tOR
x (where t is 1 or 2) and -R
y-S(O)
tN(R
x)
2 (where t is 1 or 2), where each R
x is independently hydrogen, alkyl (optionally substituted with halogen, hydroxy, methoxy, or trifluoromethyl), fluoroalkyl, cycloalkyl (optionally substituted with halogen, hydroxy, methoxy, or trifluoromethyl), cycloalkylalkyl (optionally substituted with halogen, hydroxy, methoxy, or trifluoromethyl), aryl (optionally substituted with halogen, hydroxy, methoxy, or trifluoromethyl), aralkyl (optionally substituted with halogen, hydroxy, methoxy, or trifluoromethyl), heterocyclyl (optionally substituted with halogen, hydroxy, methoxy, or trifluoromethyl), heterocyclylalkyl (optionally substituted with halogen, hydroxy, methoxy, or trifluoromethyl), heteroaryl (optionally substituted with halogen, hydroxy, methoxy, or trifluoromethyl), or heteroarylalkyl (optionally substituted with halogen, hydroxy, methoxy, or trifluoromethyl), each R
y is independently a direct bond or a straight or branched alkylene or alkenylene chain, and R
z is a straight or
WSGR Docket No.55776-726.601 branched alkylene or alkenylene chain, and where each of the above substituents is unsubstituted unless otherwise indicated. [0022] As used herein, the term “aryl” refers to an aromatic ring wherein each of the atoms forming the ring is a carbon atom. The aromatic monocyclic or multicyclic hydrocarbon ring system contains only hydrogen and carbon from five to eighteen carbon atoms, where at least one of the rings in the ring system is fully unsaturated, i.e., it contains a cyclic, delocalized (4n+2) ^–electron system in accordance with the Hückel theory. The ring system from which aryl groups are derived include, but are not limited to, groups such as benzene, fluorene, indane, indene, tetralin and naphthalene. Unless stated otherwise specifically in the specification, the term "aryl" or the prefix "ar-" (such as in "aralkyl") is meant to include aryl radicals optionally substituted by one or more substituents independently selected from alkyl, alkenyl, alkynyl, halo, fluoroalkyl, cyano, nitro, optionally substituted aryl, optionally substituted aralkyl, optionally substituted aralkenyl, optionally substituted aralkynyl, optionally substituted carbocyclyl, optionally substituted carbocyclylalkyl, optionally substituted heterocyclyl, optionally substituted heterocyclylalkyl, optionally substituted heteroaryl, optionally substituted heteroarylalkyl, -R
y-OR
x, -R
y-OC(O)-R
x, -R
y-OC(O)-OR
x, -R
y- OC(O)-N(R
x)
2, -R
y-N(R
x)
2, -R
y-C(O)R
x, -R
y-C(O)OR
x, -R
y-C(O)N(R
x)
2, -R
y-O-R
z-C(O)N(R
x)
2, -R
y- N(R
x)C(O)OR
x, -R
y-N(R
x)C(O)R
x, -R
y-N(R
x)S(O)
tR
x (where t is 1 or 2), -R
y-S(O)
tR
x (where t is 1 or 2), -R
y- S(O)tOR
x (where t is 1 or 2) and -R
y-S(O)tN(R
x)2 (where t is 1 or 2), where each R
x is independently hydrogen, alkyl (optionally substituted with halogen, hydroxy, methoxy, or trifluoromethyl), fluoroalkyl, cycloalkyl (optionally substituted with halogen, hydroxy, methoxy, or trifluoromethyl), cycloalkylalkyl (optionally substituted with halogen, hydroxy, methoxy, or trifluoromethyl), aryl (optionally substituted with halogen, hydroxy, methoxy, or trifluoromethyl), aralkyl (optionally substituted with halogen, hydroxy, methoxy, or trifluoromethyl), heterocyclyl (optionally substituted with halogen, hydroxy, methoxy, or trifluoromethyl), heterocyclylalkyl (optionally substituted with halogen, hydroxy, methoxy, or trifluoromethyl), heteroaryl (optionally substituted with halogen, hydroxy, methoxy, or trifluoromethyl), or heteroarylalkyl (optionally substituted with halogen, hydroxy, methoxy, or trifluoromethyl), each R
y is independently a direct bond or a straight or branched alkylene or alkenylene chain, and R
z is a straight or branched alkylene or alkenylene chain, and where each of the above substituents is unsubstituted unless otherwise indicated. [0023] The term “cycloalkyl” refers to a monocyclic or polycyclic aliphatic, non-aromatic radical, wherein each of the atoms forming the ring (i.e., skeletal atoms) is a carbon atom. In some embodiments, cycloalkyls are spirocyclic or bridged compounds. In some embodiments, cycloalkyls are optionally fused with an aromatic ring, and the point of attachment is at a carbon that is not an aromatic ring carbon atom. Cycloalkyl groups include groups having from 3 to 10 ring atoms. In some embodiments, cycloalkyl groups are selected from among cyclopropyl, cyclobutyl, cyclopentyl, cyclopentenyl, cyclohexyl, cyclohexenyl, cycloheptyl, cyclooctyl, spiro[2.2]pentyl, norbornyl and bicycle[1.1.1]pentyl, bicyclo[3.3.0]octane, bicyclo[4.3.0]nonane, cis-decalin, trans-decalin, bicyclo[2.1.1]hexane, bicyclo[2.2.1]heptane, bicyclo[2.2.2]octane,
WSGR Docket No.55776-726.601 bicyclo[3.2.2]nonane, and bicyclo[3.3.2]decane, adamantyl, norbornyl, and decalinyl. In some embodiments, a cycloalkyl is a C
3-C
6cycloalkyl. [0024] The term “halo” or, alternatively, “halogen” or “halide” means fluoro, chloro, bromo or iodo. In some embodiments, halo is fluoro, chloro, or bromo. In some embodiments, halo is fluoro or chloro. [0025] The term “heteroalkyl” refers to an alkyl group as defined above in which one or more skeletal carbon atoms of the alkyl are substituted with a heteroatom (with the appropriate number of substituents or valencies – for example, -CH
2- may be replaced with -NH-, -S-, or -O-). For example, each substituted carbon atom is independently substituted with a heteroatom, such as wherein the carbon is substituted with a nitrogen, oxygen, selenium, or other suitable heteroatom. In some embodiments, each substituted carbon atom is independently substituted for an oxygen, nitrogen (e.g., -NH-, -N(alkyl)-, or -N(aryl)- or having another substituent contemplated herein), or sulfur (e.g., -S-, -S(=O)-, or -S(=O)2-). In some embodiments, a heteroalkyl is attached to the rest of the molecule at a carbon atom of the heteroalkyl. In some embodiments, a heteroalkyl is attached to the rest of the molecule at a heteroatom of the heteroalkyl. In some embodiments, a heteroalkyl is a C1-C18 heteroalkyl. In some embodiments, a heteroalkyl is a C1-C12 heteroalkyl. In some embodiments, a heteroalkyl is a C1-C6 heteroalkyl. In some embodiments, a heteroalkyl is a C1-C4 heteroalkyl. In some embodiments, a heteroalkyl is or includes one or more cyclic group(s). In some embodiments, heteroalkyl includes alkylamino, alkylaminoalkyl, aminoalkyl, heterocyclyl, heterocycloalkyl, heterocycloalkyl, and heterocycloalkylalkyl, as defined herein. Unless stated otherwise specifically in the specification, a heteroalkyl group is optionally substituted as defined above for an alkyl group. In one embodiment, a heteroalkyl is a C1-C6heteroalkyl. [0026] “Heteroalkylene” refers to a divalent heteroalkyl group defined above which links one part of the molecule to another part of the molecule. Unless stated specifically otherwise, a heteroalkylene is optionally substituted, as defined above for an alkyl group. [0027] The terms “heteroaryl” or, alternatively, “heteroaromatic” refers to an aryl group that includes one or more ring heteroatoms selected from nitrogen, oxygen and sulfur. Illustrative examples of heteroaryl groups include monocyclic heteroaryls and bicyclcic heteroaryls. Monocyclic heteroaryls include pyridinyl, imidazolyl, pyrimidinyl, pyrazolyl, triazolyl, pyrazinyl, tetrazolyl, furyl, thienyl, isoxazolyl, thiazolyl, oxazolyl, isothiazolyl, pyrrolyl, pyridazinyl, triazinyl, oxadiazolyl, thiadiazolyl, and furazanyl. Bicyclic heteroaryls include indolizine, indole, benzofuran, benzothiophene, indazole, benzimidazole, purine, quinolizine, quinoline, isoquinoline, cinnoline, phthalazine, quinazoline, quinoxaline, 1,8-naphthyridine, and pteridine. In some embodiments, a heteroaryl contains 0-4 N atoms in the ring. In some embodiments, a heteroaryl contains 1-4 N atoms in the ring. In some embodiments, a heteroaryl contains 0-4 N atoms, 0-1 O atoms, and 0-1 S atoms in the ring. In some embodiments, a heteroaryl contains 1-4 N atoms, 0-1 O atoms, and 0-1 S atoms in the ring. In some embodiments, heteroaryl is a C
1-C
9heteroaryl. In some embodiments, monocyclic heteroaryl is a C
1-C
5heteroaryl. In some embodiments, monocyclic heteroaryl is a 5-membered or 6-membered heteroaryl. In some embodiments, bicyclic heteroaryl is a C
6-C
9heteroaryl. Unless stated otherwise specifically in the specification, the term "heteroaryl" is meant to include heteroaryl radicals as
WSGR Docket No.55776-726.601 defined above which are optionally substituted by one or more substituents selected from alkyl, alkenyl, alkynyl, halo, fluoroalkyl, haloalkenyl, haloalkynyl, oxo, thioxo, cyano, nitro, optionally substituted aryl, optionally substituted aralkyl, optionally substituted aralkenyl, optionally substituted aralkynyl, optionally substituted carbocyclyl, optionally substituted carbocyclylalkyl, optionally substituted heterocyclyl, optionally substituted heterocyclylalkyl, optionally substituted heteroaryl, optionally substituted heteroarylalkyl, -R
y-OR
x, -R
y-OC(O)-R
x, -R
y-OC(O)-OR
x, -R
y-OC(O)-N(R
x)
2, -R
y-N(R
x)
2, -R
y-C(O)R
x, -R
y- C(O)OR
x, -R
y-C(O)N(R
x)
2, -R
y-O-R
z-C(O)N(R
x)
2, -R
y-N(R
x)C(O)OR
x, -R
y-N(R
x)C(O)R
x, -R
y-N(R
x)S(O)
tR
x (where t is 1 or 2), -R
y-S(O)
tR
x (where t is 1 or 2), -R
y-S(O)
tOR
x (where t is 1 or 2) and -R
y-S(O)
tN(R
x)
2 (where t is 1 or 2), where each R
x is independently hydrogen, alkyl (optionally substituted with halogen, hydroxy, methoxy, or trifluoromethyl), fluoroalkyl, cycloalkyl (optionally substituted with halogen, hydroxy, methoxy, or trifluoromethyl), cycloalkylalkyl (optionally substituted with halogen, hydroxy, methoxy, or trifluoromethyl), aryl (optionally substituted with halogen, hydroxy, methoxy, or trifluoromethyl), aralkyl (optionally substituted with halogen, hydroxy, methoxy, or trifluoromethyl), heterocyclyl (optionally substituted with halogen, hydroxy, methoxy, or trifluoromethyl), heterocyclylalkyl (optionally substituted with halogen, hydroxy, methoxy, or trifluoromethyl), heteroaryl (optionally substituted with halogen, hydroxy, methoxy, or trifluoromethyl), or heteroarylalkyl (optionally substituted with halogen, hydroxy, methoxy, or trifluoromethyl), each R
y is independently a direct bond or a straight or branched alkylene or alkenylene chain, and R
z is a straight or branched alkylene or alkenylene chain, and where each of the above substituents is unsubstituted unless otherwise indicated. [0028] A “heterocycloalkyl” or “heteroalicyclic” group refers to a cycloalkyl group that includes at least one heteroatom selected from nitrogen, oxygen and sulfur. In some embodiments, a heterocycloalkyl is fused with an aryl or heteroaryl. In some embodiments, the heterocycloalkyl is oxazolidinonyl, pyrrolidinyl, tetrahydrofuranyl, tetrahydrothienyl, tetrahydropyranyl, tetrahydrothiopyranyl, piperidinyl, morpholinyl, thiomorpholinyl, piperazinyl, piperidin-2-onyl, pyrrolidine-2,5-dithionyl, pyrrolidine-2,5-dionyl, pyrrolidinonyl, imidazolidinyl, imidazolidin-2-onyl, or thiazolidin-2-onyl. The term heteroalicyclic also includes all ring forms of the carbohydrates, including but not limited to the monosaccharides, the disaccharides and the oligosaccharides. In one embodiment, a heterocycloalkyl is a C2-C10heterocycloalkyl. In another embodiment, a heterocycloalkyl is a C4-C10heterocycloalkyl. In some embodiments, a heterocycloalkyl contains 0-2 N atoms in the ring. In some embodiments, a heterocycloalkyl contains 0-2 N atoms, 0-2 O atoms, and 0-1 S atoms in the ring. [0029] The term “bond” or “single bond” refers to a chemical bond between two atoms, or two moieties when the atoms joined by the bond are considered to be part of larger substructure. In one embodiment, when a group described herein is a bond, the referenced group is absent thereby allowing a bond to be formed between the remaining identified groups. [0030] The term “moiety” refers to a specific segment or functional group of a molecule. Chemical moieties are often recognized chemical entities embedded in or appended to a molecule.
WSGR Docket No.55776-726.601 [0031] In general, optionally substituted groups are each independently substituted or unsubstituted. Each recitation of an optionally substituted group provided herein, unless otherwise stated, includes an independent and explicit recitation of both an unsubstituted group and a substituted group (e.g., substituted in certain embodiments, and unsubstituted in certain other embodiments). Unless otherwise stated, a substituted group provided herein (e.g., substituted alkyl) is substituted by one or more substituent, each substituent being independently selected from the group consisting of: halo, cyano, nitro, oxo, thioxo, imino, oximo, trimethylsilanyl, -OR
x, -SR
x, -OC(O)-R
x, -N(R
x)
2, -C(O)R
x, -C(O)OR
x, -C(O)N(R
x)
2, - N(R
x)C(O)OR
x, -OC(O)-N(R
x)
2, -N(R
x)C(O)R
x, -N(R
x)S(O)
tR
x (where t is 1 or 2), -S(O)
tOR
x (where t is 1 or 2), -S(O)tR
x (where t is 1 or 2) and -S(O)tN(R
x)2 (where t is 1 or 2) where each R
x is independently hydrogen, alkyl (optionally substituted with halogen, hydroxy, methoxy, or trifluoromethyl), fluoroalkyl, carbocyclyl (optionally substituted with halogen, hydroxy, methoxy, or trifluoromethyl), carbocyclylalkyl (optionally substituted with halogen, hydroxy, methoxy, or trifluoromethyl), aryl (optionally substituted with halogen, hydroxy, methoxy, or trifluoromethyl), aralkyl (optionally substituted with halogen, hydroxy, methoxy, or trifluoromethyl), heterocyclyl (optionally substituted with halogen, hydroxy, methoxy, or trifluoromethyl), heterocyclylalkyl (optionally substituted with halogen, hydroxy, methoxy, or trifluoromethyl), heteroaryl (optionally substituted with halogen, hydroxy, methoxy, or trifluoromethyl), or heteroarylalkyl (optionally substituted with halogen, hydroxy, methoxy, or trifluoromethyl). In some other embodiments, optional substituents are independently selected from halogen, -CN, -NH2, -NH(CH3), - N(CH3)2, -OH, -CO2H, -CO2(C1-C4alkyl), -C(=O)NH2, -C(=O)NH(C1-C4alkyl), -C(=O)N(C1-C4alkyl)2, - S(=O)2NH2, -S(=O)2NH(C1-C4alkyl), -S(=O)2N(C1-C4alkyl)2, C1-C4alkyl, C3-C6cycloalkyl, C1-C4fluoroalkyl, C1-C4heteroalkyl, C1-C4alkoxy, C1-C4fluoroalkoxy, -SC1-C4alkyl, -S(=O)C1-C4alkyl, and -S(=O)2C1-C4alkyl. In some embodiments, optional substituents are independently selected from halogen, -CN, -NH2, -OH, - NH(CH3), -N(CH3)2, -CH3, -CH2CH3, -CF3, -OCH3, and -OCF3. In some embodiments, substituted groups are substituted with one or two of the preceding groups. In some embodiments, an optional substituent on an aliphatic carbon atom (acyclic or cyclic) includes oxo (=O). [0032] The term “acceptable” with respect to a formulation, composition or ingredient, as used herein, means having no persistent detrimental effect on the general health of the subject being treated. [0033] The term “modulate” as used herein, means to interact with a target either directly or indirectly so as to alter the activity of the target, including, by way of example only, to enhance the activity of the target, to inhibit the activity of the target, to limit the activity of the target, or to extend the activity of the target. In some embodiments, “modulate” means to interact with a target either directly or indirectly so as to decrease or inhibit receptor activity. In some embodiments. modulation is an increase or decrease in the amount, quality, or effect of a particular activity, function or molecule. By way of illustration and not limitation, agonists, partial agonists, antagonists, and allosteric modulators (e.g., a positive allosteric modulator) of a G protein-coupled receptor are modulators of the receptor. [0034] The term “modulator” as used herein, refers to a molecule that interacts with a target either directly or indirectly. The interactions include, but are not limited to, the interactions of an agonist, partial agonist,
WSGR Docket No.55776-726.601 an inverse agonist, antagonist, or combinations thereof. In some embodiments, a modulator is an antagonist. Receptor antagonists are inhibitors of receptor activity. Antagonists mimic ligands that bind to a receptor and prevent receptor activation by a natural ligand. Preventing activation may have many effects. If a natural agonist binding to a receptor leads to an increase in cellular function, an antagonist that binds and blocks this receptor decreases the function of the receptor. [0035] The term “agonism,” as used herein, generally refers to the activation of a receptor or enzyme by a modulator, or agonist, to produce a biological response. [0036] The term “agonist,” as used herein, generally refers to a modulator that binds to a receptor or enzyme and activates the receptor to produce a biological response. In some embodiments, the term “agonist” includes full agonists or partial agonists. “Full agonist” refers to a modulator that binds to and activates a receptor with the maximum response that an agonist can elicit at the receptor. “Partial agonist” refers to a modulator that binds to and activates a given receptor, but has partial efficacy, that is, less than the maximal response, at the receptor relative to a full agonist. [0037] The term “positive allosteric modulator,” as used herein, generally refers to a modulator that binds to a site distinct from the orthosteric binding site and enhances or amplifies the effect of an agonist. [0038] The term “antagonism,” as used herein, generally refers to the inactivation of a receptor or enzyme by a modulator, or antagonist. Antagonism of a receptor, for example, is when a molecule binds to the receptor and blocks function of the receptor. [0039] The term “antagonist” or “neutral antagonist,” as used herein, generally refers to a modulator that binds to a receptor or enzyme and blocks a biological response. An antagonist may have no activity in the absence of an agonist or inverse agonist but can block the activity of either, causing no change in the biological response. [0040] The terms "administer," "administering", "administration," and the like, as used herein, refer to the methods that may be used to enable delivery of compounds or compositions to the desired site of biological action. These methods include, but are not limited to oral routes, intraduodenal routes, parenteral injection (including intravenous, subcutaneous, intraperitoneal, intramuscular, intravascular or infusion), topical and rectal administration. Those of skill in the art are familiar with administration techniques that can be employed with the compounds and methods described herein. In some embodiments, the compounds and compositions described herein are administered orally. [0041] The terms “effective amount” or “therapeutically effective amount,” as used herein, refer to a sufficient amount of an agent or a compound being administered, which will relieve to some extent one or more of the symptoms of the disease or condition being treated. The result includes reduction and/or alleviation of the signs, symptoms, or causes of a disease, or any other desired alteration of a biological system. For example, an “effective amount” for therapeutic uses is the amount of the composition comprising a compound as disclosed herein required to provide a clinically significant decrease in disease symptoms. An appropriate “effective” amount in any individual case is optionally determined using techniques, such as a dose escalation study.
WSGR Docket No.55776-726.601 [0042] The term “subject” or “patient” encompasses mammals. Examples of mammals include, but are not limited to, any member of the Mammalian class: humans, non-human primates such as chimpanzees, and other apes and monkey species; farm animals such as cattle, horses, sheep, goats, swine; domestic animals such as rabbits, dogs, and cats; laboratory animals including rodents, such as rats, mice and guinea pigs, and the like. In one embodiment, the mammal is a human. [0043] The terms “treat,” “treating” or “treatment,” as used herein, include alleviating, abating or ameliorating at least one symptom of a disease or condition, preventing additional symptoms, inhibiting the disease or condition, e.g., arresting the development of the disease or condition, relieving the disease or condition, causing regression of the disease or condition, relieving a condition caused by the disease or condition, or stopping the symptoms of the disease or condition either prophylactically and/or therapeutically. [0044] The term “pharmaceutically acceptable,” as used herein, generally refers a material, such as a carrier or diluent, which does not abrogate the biological activity or properties of the compound, and is relatively nontoxic, i.e., the material is administered to an individual without causing undesirable biological effects or interacting in a deleterious manner with any of the components of the composition in which it is contained. [0045] The term “pharmaceutically acceptable salt,” as used herein, generally refers to a form of a therapeutically active agent that consists of a cationic form of the therapeutically active agent in combination with a suitable anion, or in alternative embodiments, an anionic form of the therapeutically active agent in combination with a suitable cation. Handbook of Pharmaceutical Salts: Properties, Selection and Use. International Union of Pure and Applied Chemistry, Wiley-VCH 2002. S.M. Berge, L.D. Bighley, D.C. Monkhouse, J. Pharm. Sci.1977, 66, 1-19. P. H. Stahl and C. G. Wermuth, editors, Handbook of Pharmaceutical Salts: Properties, Selection and Use, Weinheim/Zürich:Wiley-VCH/VHCA, 2002. Pharmaceutical salts typically are more soluble and more rapidly soluble in stomach and intestinal juices than non-ionic species and so are useful in solid dosage forms. Furthermore, because their solubility often is a function of pH, selective dissolution in one or another part of the digestive tract is possible and this capability can be manipulated as one aspect of delayed and sustained release behaviors. Also, because the salt-forming molecule can be in equilibrium with a neutral form, passage through biological membranes can be adjusted. Provided herein are non-hallucinogenic compounds that promote neuronal growth and/or improve neuronal structure. Compounds [0046] In one aspect, disclosed herein is a compound of Formula (I), or a pharmaceutically acceptable salt, pharmaceutically acceptable solvate, diastereomeric mixture, individual enantiomer, or isotopically enriched form thereof:
WSGR Docket No.55776-726.601

wherein: each R
3, R
4, and R
5 is independently hydrogen, C
1-C
6alkyl, C
1-C
6haloalkyl, C
1-C
6hydroxyalkyl, C
1- C
6aminoalkyl, C
1-C
6heteroalkyl, halogen, -CN, -NO
2, -OR
a, -SR
a, -NR
cR
d, -S(=O)R
b, -S(=O)
2R
b, - S(=O)
2NR
cR
d, -NR
bS(=O)
2R
b, -NR
bS(=O)
2NR
cR
d, -C(=O)R
b, -C(=O)OR
b, -OC(=O)R
b, - OC(=O)OR
b, -OC(=O)NR
cR
d, -NR
bC(=O)R
b, -NR
bC(=O)OR
b, -NR
bC(=O)NR
cR
d, -C(=O)NR
cR
d, - P(=O)(OR
c)(OR
d), -P(=O)R
cR
d, aryl, heteroaryl, cycloalkyl, heterocycloalkyl, C
1-C
6alkyl(cycloalkyl), C
1-C
6alkyl(heterocycloalkyl), C
1-C
6alkyl(aryl), or C
1-C
6alkyl(heteroaryl), wherein each of the alkyl, heteroalkyl, aryl, heteroaryl, cycloalkyl, and heterocycloalkyl is optionally substituted with one or more substituents selected from C
1-C
3alkyl, C
1-C
3haloalkyl, C
1- C
3alkoxy, halogen, -OH, -CN, and =O; R
8 is hydrogen or C
1-C
6alkyl; R
9 is hydrogen or C
1-C
6alkyl; or R
8 and R
9 are taken together to form a heterocycloalkyl, wherein the heterocycloalkyl is optionally substituted with one or more substituents selected from C1-C3alkyl, C1-C3haloalkyl, C1-C3alkoxy, halogen, -OH, and =O; each R
a is independently hydrogen, C1-C6alkyl, C1-C6haloalkyl, C1-C6heteroalkyl, aryl, C1- C3alkyl(phenyl), C3-C6cycloalkyl, 5- to 6- membered heteroaryl, C1-C3alkyl(5- to 6- membered heteroaryl), or 4- to 6- membered heterocycloalkyl, wherein each alkyl, heteroalkyl, aryl, cycloalkyl, heteroaryl, and heterocycloalkyl is optionally substituted with one or more substituents selected from C1-C3alkyl, C1-C3haloalkyl, C1-C3alkoxy, halogen, -OH, -CN, and =O; each R
b is independently hydrogen or C1-C6alkyl; each R
c and R
d is independently hydrogen or C1-C6alkyl; or R
c and R
d are taken together to form a 4- to 6- membered heterocycloalkyl, wherein the heterocycloalkyl is optionally substituted with one or more substituents selected from C1-C3alkyl, C1-C3haloalkyl, C1- C3alkoxy, halogen, -OH, -CN, and =O; wherein: at least two of R
3, R
4, and R
5 are not hydrogen.
WSGR Docket No.55776-726.601 [0047] In some embodiments, the compound of Formula (I) has the structure of Formula (I-A-8), or a pharmaceutically acceptable salt, pharmaceutically acceptable solvate, diastereomeric mixture, individual enantiomer, or isotopically enriched form thereof:
Formula (I-A-8). [0048] In some embodiments, the compound of Formula (I) has the structure of Formula (I-A-9), or a pharmaceutically acceptable salt, pharmaceutically acceptable solvate, diastereomeric mixture, individual enantiomer, or isotopically enriched form thereof:
Formula (I-A-9). [0049] In some embodiments, the compound of Formula (I) has the structure of Formula (I-A-10), or a pharmaceutically acceptable salt, pharmaceutically acceptable solvate, diastereomeric mixture, individual enantiomer, or isotopically enriched form thereof:
Formula (I-A-10). [0050] In some embodiments of Formula (I), (I-A-8), (I-A-9), or (I-A-10), each R
3, R
4, and R
5, when present, is independently hydrogen, C
1-C
6alkyl, C
1-C
6haloalkyl, C
1-C
6hydroxyalkyl, C
1-C
6aminoalkyl, C
1- C
6heteroalkyl, halogen, -CN, -NO
2, -OR
a, -SR
a, -NR
cR
d, -S(=O)R
b, -S(=O)
2R
b, -S(=O)
2NR
cR
d, - NR
bS(=O)
2R
b, -NR
bS(=O)
2NR
cR
d, -C(=O)R
b, -C(=O)OR
b, -OC(=O)R
b, -OC(=O)OR
b, -OC(=O)NR
cR
d, - NR
bC(=O)R
b, -NR
bC(=O)OR
b, -NR
bC(=O)NR
cR
d, -C(=O)NR
cR
d, -P(=O)(OR
c)(OR
d), -P(=O)R
cR
d, C
6-
WSGR Docket No.55776-726.601 C
10aryl, 5-10 membered heteroaryl, C
3-C
7cycloalkyl, 3- to 10- membered heterocycloalkyl, C
1-C
6alkyl(C
3- C
7cycloalkyl), C
1-C
6alkyl(3- to 10- membered heterocycloalkyl), C
1-C
6alkyl(C
6-C
10aryl), or C
1-C
6alkyl(heteroaryl), wherein each of the alkyl, heteroalkyl, aryl, heteroaryl, cycloalkyl, and heterocycloalkyl is optionally substituted with one or more substituents selected from C
1-C
3alkyl, C
1- C
3haloalkyl, C
1-C
3alkoxy, halogen, -OH, -CN, and =O. In some embodiments, each R
3, R
4, and R
5 is independently hydrogen, C
1-C
6alkyl, C
1-C
6heteroalkyl, halogen, C
6-C
10aryl, or C
3-C
7cycloalkyl, wherein each of the alkyl, heteroalkyl, aryl, and cycloalkyl is optionally substituted with one or more substituents selected from halogen, C
1-C
3alkyl, C
1-C
3alkoxy, halogen, -OH, and -CN. In some embodiments, each R
3, R
4, and R
5 is independently hydrogen, C1-C6alkyl, C1-C6heteroalkyl, -F, -Cl, -Br, phenyl, cyclopropyl, or cyclobutyl, wherein each of the alkyl, heteroalkyl, phenyl, cyclopropyl, and cyclobutyl is optionally substituted with one or more substituents selected from C1-C3alkyl, C1-C3alkoxy, -F, -Cl, -Br, and -CN. In some embodiments, each R
3, R
4, and R
5 is independently hydrogen, C1-C6alkyl, C1-C6heteroalkyl, -F, -Cl, - Br, phenyl, cyclopropyl, or cyclobutyl, wherein each of the alkyl, heteroalkyl, phenyl, cyclopropyl, and cyclobutyl is optionally substituted with one or more substituents selected from methyl, -O-CH3, -F, -Cl, -Br, and -CN. In some embodiments, each R
3, R
4, and R
5 is independently hydrogen, -CH3, -F, -Cl, or -Br
[0051] In some embodiments of Formula (I), (I-A-2), (I-A-5), (I-A-8), (I-A-9), (I-A-11), (I-A-14), (I-A-15), (I-A-17), (I-A-18), or (I-A-20), R
3 is hydrogen, C
1-C
6alkyl, C
1-C
6heteroalkyl, -F, -Cl, -Br, or phenyl, wherein each of the alkyl, heteroalkyl, and phenyl is optionally substituted with one or more substituents selected from C1-C3alkyl, C1-C3alkoxy, -F, -Cl, -Br, and -CN. In some embodiments, R
3 is hydrogen. In some embodiments, R
3 is C1-C6alkyl, C1-C6heteroalkyl, -F, -Cl, -Br, or phenyl, wherein each of the alkyl, heteroalkyl, and phenyl is optionally substituted with one or more substituents selected from C1-C3alkyl, C1- C3alkoxy, -F, -Cl, -Br, and -CN. In some embodiments, R
3 is C1-C6alkyl, C1-C6heteroalkyl, -F, -Cl, -Br, or phenyl, wherein each of the alkyl, heteroalkyl, and phenyl is optionally substituted with one or more substituents selected from methyl, -O-CH3, -F, -Cl, -Br, and -CN. [0052] In some embodiments of Formula (I), (I-A-3), (I-A-6), (I-A-8), (I-A-10), (I-A-12), (I-A-14), (I-A- 16), (I-A-17), (I-A-19), or (I-A-20), R
4 is hydrogen, C1-C6alkyl, C1-C6heteroalkyl, -F, -Cl, -Br, or phenyl, wherein each of the alkyl, heteroalkyl, and phenyl is optionally substituted with one or more substituents selected from C1-C3alkyl, C1-C3alkoxy, -F, -Cl, -Br, and -CN. In some embodiments, R
4 is hydrogen. In some embodiments, R
4 is C1-C6alkyl, C1-C6heteroalkyl, -F, -Cl, -Br, or phenyl, wherein each of the alkyl, heteroalkyl, and phenyl is optionally substituted with one or more substituents selected from C1-C3alkyl, C1- C3alkoxy, -F, -Cl, -Br, and -CN. In some embodiments, R
4 is C1-C6alkyl, C1-C6heteroalkyl, -F, -Cl, -Br, or phenyl, wherein each of the alkyl, heteroalkyl, and phenyl is optionally substituted with one or more substituents selected from methyl, -O-CH3, -F, -Cl, -Br, and -CN.
WSGR Docket No.55776-726.601 [0053] In some embodiments of Formula (I), (I-A-4), (I-A-7), (I-A-9), (I-A-10), (I-A-13), (I-A-15), (I-A- 16), (I-A-18), (I-A-19), or (I-A-20), R
5 is hydrogen, C
1-C
6alkyl, C
1-C
6heteroalkyl, -F, -Cl, -Br, or phenyl, wherein each of the alkyl, heteroalkyl, and phenyl is optionally substituted with one or more substituents selected from C
1-C
3alkyl, C
1-C
3alkoxy, -F, -Cl, -Br, and -CN. In some embodiments, R
5 is hydrogen. In some embodiments, R
5 is C
1-C
6alkyl, C
1-C
6heteroalkyl, -F, -Cl, -Br, or phenyl, wherein each of the alkyl, heteroalkyl, and phenyl is optionally substituted with one or more substituents selected from C
1-C
3alkyl, C
1- C
3alkoxy, -F, -Cl, -Br, and -CN. In some embodiments, R
5is C
1-C
6alkyl, C
1-C
6heteroalkyl, -F, -Cl, -Br, or phenyl, wherein each of the alkyl, heteroalkyl, and phenyl is optionally substituted with one or more substituents selected from methyl, -O-CH3, -F, -Cl, -Br, and -CN. [0054] In some embodiments of Formula (I), (I-A-8), (I-A-9), or (I-A-10),R
8 is hydrogen or C1-C6alkyl. In some embodiments, R
8 is hydrogen or C1-C3alkyl. In some embodiments, R
8 is hydrogen. In some embodiments, R
8 is C1-C3alkyl. [0055] In some embodiments of Formula (I), (I-A-8), (I-A-9), or (I-A-10), R
9 is hydrogen or C1-C3alkyl. In some embodiments, R
9 is hydrogen. In some embodiments, R
9 is -CH3. In some embodiments, R
9 is - CH2CH3. [0056] In some embodiments of Formula (I), (I-A-8), (I-A-9), or (I-A-10), R
8 and R
9 are taken together to form a heterocycloalkyl, wherein the heterocycloalkyl is optionally substituted with one or more substituents selected from C1-C3alkyl, C1-C3haloalkyl, C1-C3alkoxy, halogen, -OH, and =O. In some embodiments, the heterocycloalkyl is a 5- to 6- membered heterocycloalkyl optionally substituted with one or more substituents selected from C1-C3alkyl, C1-C3haloalkyl, C1-C3alkoxy, halogen, -OH, and =O. In some embodiments, R
8 and R
9 are taken together to form a pyrrolidinyl, an imidazolidinyl, a piperidinyl, a piperazinyl, or a morpholino, each of which is optionally substituted with one or more substituents selected from C1-C3alkyl, C1-C3haloalkyl, C1-C3alkoxy, halogen, -OH, and =O. In some embodiments, R
8 and R
9 are taken together to form a pyrrolidinyl. In some embodiments, R
8 and R
9 are taken together to form an imidazolidinyl. In some embodiments, R
8 and R
9 are taken together to form a piperidinyl. In some embodiments, R
8 and R
9 are taken together to form a piperazinyl. In some embodiments, R
8 and R
8 are taken together to form a morpholino. [0057] In some embodiments, a compound disclosed herein, or a pharmaceutically acceptable salt, pharmaceutically acceptable solvate, diastereomeric mixture, individual enantiomer, or isotopically enriched
, , or . [0058] In some embodiments, a compound disclosed herein, or a pharmaceutically acceptable salt, pharmaceutically acceptable solvate, diastereomeric mixture, individual enantiomer, or isotopically enriched
WSGR Docket No.55776-726.601 , ,

[0059] In another aspect, disclose herein is a compound disclosed herein, or a pharmaceutically acceptable salt, pharmaceutically acceptable solvate, diastereomeric mixture, individual enantiomer, or isotopically enriched form thereof, for use as medicine. [0060] Provided in some embodiments herein is a compound of Formula (I), (I-A-8), (I-A-9), or (I-A-10), or a pharmaceutically acceptable salt, pharmaceutically acceptable solvate, diastereomeric mixture, individual enantiomer, or isotopically enriched form thereof, having a structure provided in Table 1. Table 1 Compound Structure Chemical Name 8,9-difluoro-5,6-dihydro-4H-pyrrolo[3,2,1- 9 ij]quinolin-5-amine
WSGR Docket No.55776-726.601 (R)-8,9-difluoro-5,6-dihydro-4H- 10 pyrrolo[3,2,1-ij]quinolin-5-amine (S)-8,9-difluoro-5,6-dihydro-4H- 11 pyrrolo[3,2,1-ij]quinolin-5-amine 8,9-difluoro-N,N-dimethyl-5,6-dihydro- 12 4H-pyrrolo[3,2,1-ij]quinolin-5-amine (R)-8,9-difluoro-N,N-dimethyl-5,6- 13 dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5- amine (S)-8,9-difluoro-N,N-dimethyl-5,6- 14 dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5- amine (S)-9-chloro-8-fluoro-N-methyl-5,6- 61 dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5- amine (R)-9-chloro-8-fluoro-N-methyl-5,6- 62 dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5- amine
WSGR Docket No.55776-726.601 (R)-8,9-difluoro-N-methyl-5,6-dihydro- 63 4H-pyrrolo[3,2,1-ij]quinolin-5-amine (S)-9-chloro-8-fluoro-N,N-dimethyl-5,6- 64 dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5- amine (R)-9-chloro-8-fluoro-N,N-dimethyl-5,6- 65 dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5- amine (S)-8,9-difluoro-N-methyl-5,6-dihydro- 66 4H-pyrrolo[3,2,1-ij]quinolin-5-amine 8-fluoro-N,9-dimethyl-5,6-dihydro-4H- 67 pyrrolo[3,2,1-ij]quinolin-5-amine 9-chloro-8-fluoro-N-methyl-5,6-dihydro- 68 4H-pyrrolo[3,2,1-ij]quinolin-5-amine 8-chloro-9-fluoro-N-methyl-5,6-dihydro- 69 4H-pyrrolo[3,2,1-ij]quinolin-5-amine
WSGR Docket No.55776-726.601 8-chloro-9-fluoro-N,N-dimethyl-5,6- 70 dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5- amine 9-chloro-8-fluoro-N,N-dimethyl-5,6- 71 dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5- amine 8,9-difluoro-N-methyl-5,6-dihydro-4H- 72 pyrrolo[3,2,1-ij]quinolin-5-amine 8-fluoro-N,N,9-trimethyl-5,6-dihydro-4H- 73 pyrrolo[3,2,1-ij]quinolin-5-amine [0061] Any combination of the groups described above for the various variables is contemplated herein. Throughout the specification, groups and substituents thereof are chosen by one skilled in the field to provide stable moieties and compounds. Pharmaceutical Compositions [0062] In another aspect, disclose herein is a pharmaceutical composition comprising the compound of Formula (I), (I-A-8), (I-A-9), or (I-A-10), or a pharmaceutically acceptable salt, pharmaceutically acceptable solvate, diastereomeric mixture, individual enantiomer, or isotopically enriched form thereof. [0063] In some embodiments, the pharmaceutical composition further comprises at least one pharmaceutically acceptable excipient. [0064] In some embodiments, disclosed herein is a pharmaceutical composition comprising a compound provided herein (e.g., a compound having a structure represented by Formula (I), (I-A-8), (I-A-9), or (I-A- 10), for example any compound described in Table 1, and a pharmaceutically acceptable salt, pharmaceutically acceptable solvate, diastereomeric mixture, individual enantiomer, or isotopically enriched form thereof. In some embodiments, the pharmaceutical composition further comprises at least one pharmaceutically acceptable excipient.
WSGR Docket No.55776-726.601 [0065] In some embodiments, the compounds described herein are formulated into pharmaceutical compositions. The pharmaceutical compositions are formulated in a conventional manner using one or more pharmaceutically acceptable inactive ingredients that facilitate processing of the active compounds into preparations that are used pharmaceutically. Proper formulation is dependent upon the route of administration chosen. A summary of pharmaceutical compositions described herein is found, for example, in Remington: The Science and Practice of Pharmacy, Nineteenth Ed (Easton, Pa.: Mack Publishing Company, 1995); Hoover, John E., Remington’s Pharmaceutical Sciences, Mack Publishing Co., Easton, Pennsylvania 1975; Liberman, H.A. and Lachman, L., Eds., Pharmaceutical Dosage Forms, Marcel Decker, New York, N.Y., 1980; and Pharmaceutical Dosage Forms and Drug Delivery Systems, Seventh Ed. (Lippincott Williams & Wilkins1999), herein incorporated by reference for such disclosure. [0066] In some embodiments, the compounds described herein are administered either alone or in combination with pharmaceutically acceptable carriers, excipients or diluents, in a pharmaceutical composition. Administration of the compounds and compositions described herein can be affected by any method that enables delivery of the compounds to the site of action. These methods include, though are not limited to delivery via enteral routes (including oral, gastric or duodenal feeding tube, rectal suppository and rectal enema), parenteral routes (injection or infusion, including intraarterial, intracardiac, intradermal, intraduodenal, intramedullary, intramuscular, intraosseous, intraperitoneal, intrathecal, intravascular, intravenous, intravitreal, epidural and subcutaneous), inhalational, transdermal, transmucosal, sublingual, buccal and topical (including epicutaneous, dermal, enema, eye drops, ear drops, intranasal, vaginal) administration, although the most suitable route may depend upon for example the condition and disorder of the recipient. By way of example only, compounds described herein can be administered locally to the area in need of treatment, by for example, local infusion during surgery, topical application such as creams or ointments, injection, catheter, or implant. The administration can also be by direct injection at the site of a diseased tissue or organ. [0067] In some embodiments, pharmaceutical compositions suitable for oral administration are presented as discrete units such as capsules, cachets or tablets each containing a predetermined amount of the active ingredient; as a powder or granules; as a solution or a suspension in an aqueous liquid or a non-aqueous liquid; or as an oil-in-water liquid emulsion or a water-in-oil liquid emulsion. In some embodiments, the active ingredient is presented as a bolus, electuary or paste. [0068] The pharmaceutical compositions which can be used orally include tablets, push-fit capsules made of gelatin, as well as soft, sealed capsules made of gelatin and a plasticizer, such as glycerol or sorbitol. Tablets may be made by compression or molding, optionally with one or more accessory ingredients. Compressed tablets may be prepared by compressing in a suitable machine the active ingredient in a free- flowing form such as a powder or granules, optionally mixed with binders, inert diluents, or lubricating, surface active or dispersing agents. Molded tablets may be made by molding in a suitable machine a mixture of the powdered compound moistened with an inert liquid diluent. In some embodiments, the tablets are coated or scored and are formulated so as to provide slow or controlled release of the active ingredient
WSGR Docket No.55776-726.601 therein. All formulations for oral administration should be in dosages suitable for such administration. The push-fit capsules can contain the active ingredients in admixture with filler such as lactose, binders such as starches, and/or lubricants such as talc or magnesium stearate and, optionally, stabilizers. In soft capsules, the active compounds may be dissolved or suspended in suitable liquids, such as fatty oils, liquid paraffin, or liquid polyethylene glycols. In some embodiments, stabilizers are added. Dragee cores are provided with suitable coatings. For this purpose, concentrated sugar solutions may be used, which may optionally contain gum arabic, talc, polyvinyl pyrrolidone, carbopol gel, polyethylene glycol, and/or titanium dioxide, lacquer solutions, and suitable organic solvents or solvent mixtures. Dyestuffs or pigments may be added to the tablets or Dragee coatings for identification or to characterize different combinations of active compound doses. [0069] In some embodiments, pharmaceutical compositions are formulated for parenteral administration by injection, e.g., by bolus injection or continuous infusion. Formulations for injection may be presented in unit dosage form, e.g., in ampoules or in multi-dose containers, with an added preservative. The compositions may take such forms as suspensions, solutions or emulsions in oily or aqueous vehicles, and may contain formulatory agents such as suspending, stabilizing and/or dispersing agents. The compositions may be presented in unit-dose or multi-dose containers, for example sealed ampoules and vials, and may be stored in powder form or in a freeze-dried (lyophilized) condition requiring only the addition of the sterile liquid carrier, for example, saline or sterile pyrogen-free water, immediately prior to use. Extemporaneous injection solutions and suspensions may be prepared from sterile powders, granules and tablets of the kind previously described. [0070] The pharmaceutical compositions for parenteral administration include aqueous and non-aqueous (oily) sterile injection solutions of the active compounds which may contain antioxidants, buffers, bacteriostats and solutes which render the formulation isotonic with the blood of the intended recipient; and aqueous and non-aqueous sterile suspensions which may include suspending agents and thickening agents. Suitable lipophilic solvents or vehicles include fatty oils such as sesame oil, or synthetic fatty acid esters, such as ethyl oleate or triglycerides, or liposomes. Aqueous injection suspensions may contain substances which increase the viscosity of the suspension, such as sodium carboxymethyl cellulose, sorbitol, or dextran. Optionally, the suspension may also contain suitable stabilizers or agents which increase the solubility of the compounds to allow for the preparation of highly concentrated solutions. [0071] It should be understood that in addition to the ingredients particularly mentioned above, the compounds and pharmaceutical compositions described herein may include other agents conventional in the art having regard to the type of formulation in question, for example those suitable for oral administration may include flavoring agents. Further Forms of Compounds [0072] In one embodiment, compounds described herein are in the form of pharmaceutically acceptable salts. In some embodiments, any compound provided herein is a pharmaceutically acceptable salt, such as, for example, any salt described herein.
WSGR Docket No.55776-726.601 [0073] As well, active metabolites of these compounds having the same type of activity are included in the scope of the present disclosure. In addition, the compounds described herein can exist in unsolvated as well as solvated forms with pharmaceutically acceptable solvents such as water, ethanol, and the like. The solvated forms of the compounds presented herein are also considered to be disclosed herein. [0074] In some embodiments, pharmaceutically acceptable salts are obtained by reacting a compound Formula (I), (I), (I-A-8), (I-A-9), or (I-A-10), for example any compound described in Table 1, with an acid. In some embodiments, the compound of Formula (I), (I-A-8), (I-A-9), or (I-A-10), for example any compound described in Table 1, (i.e., free base form) is basic and is reacted with an organic acid or an inorganic acid. Inorganic acids include, but are not limited to, hydrochloric acid, hydrobromic acid, sulfuric acid, phosphoric acid, nitric acid, and metaphosphoric acid. Organic acids include, but are not limited to, 1- hydroxy-2-naphthoic acid; 2,2-dichloroacetic acid; 2-hydroxyethanesulfonic acid; 2-oxoglutaric acid; 4- acetamidobenzoic acid; 4-aminosalicylic acid; acetic acid; adipic acid; ascorbic acid (L); aspartic acid (L); benzenesulfonic acid; benzoic acid; camphoric acid (+); camphor-10-sulfonic acid (+); capric acid (decanoic acid); caproic acid (hexanoic acid); caprylic acid (octanoic acid); carbonic acid; cinnamic acid; citric acid; cyclamic acid; dodecylsulfuric acid; ethane-1,2-disulfonic acid; ethanesulfonic acid; formic acid; fumaric acid; galactaric acid; gentisic acid; glucoheptonic acid (D); gluconic acid (D); glucuronic acid (D); glutamic acid; glutaric acid; glycerophosphoric acid; glycolic acid; hippuric acid; isobutyric acid; lactic acid (DL); lactobionic acid; lauric acid; maleic acid; malic acid (- L); malonic acid; mandelic acid (DL); methanesulfonic acid; naphthalene-1,5-disulfonic acid; naphthalene-2-sulfonic acid; nicotinic acid; oleic acid; oxalic acid; palmitic acid; pamoic acid; phosphoric acid; proprionic acid; pyroglutamic acid (- L); salicylic acid; sebacic acid; stearic acid; succinic acid; sulfuric acid; tartaric acid (+ L); thiocyanic acid; toluenesulfonic acid (p); and undecylenic acid. [0075] In some embodiments, pharmaceutically acceptable salts are obtained by reacting a compound represented by the structure of Formula (I), (I-A-8), (I-A-9), or (I-A-10), for example any compound described in Table 1,with a base. In some embodiments, the compound of represented by the structure of Formula (I), (I-A-8), (I-A-9), or (I-A-10), for example any compound described in Table 1 is acidic and is reacted with a base. In such situations, an acidic proton of the compound represented by the structure of Formula (I), (I-A-8), (I-A-9), or (I-A-10),for example any compound described in Table 1, is replaced by a metal ion, e.g., lithium, sodium, potassium, magnesium, calcium, or an aluminum ion. In some cases, compounds described herein coordinate with an organic base, such as, but not limited to, ethanolamine, diethanolamine, triethanolamine, tromethamine, meglumine, N-methylglucamine, dicyclohexylamine, tris(hydroxymethyl)methylamine. In other cases, compounds described herein form salts with amino acids such as, but not limited to, arginine, lysine, and the like. Acceptable inorganic bases used to form salts with compounds that include an acidic proton, include, but are not limited to, aluminum hydroxide, calcium hydroxide, potassium hydroxide, sodium carbonate, potassium carbonate, sodium hydroxide, lithium hydroxide, and the like. In some embodiments, the compounds provided herein are prepared as a sodium
WSGR Docket No.55776-726.601 salt, calcium salt, potassium salt, magnesium salt, meglumine salt, N-methylglucamine salt or ammonium salt. [0076] It should be understood that a reference to a pharmaceutically acceptable salt includes the solvent addition forms. In some embodiments, solvates contain either stoichiometric or non-stoichiometric amounts of a solvent, and are formed during the process of crystallization with pharmaceutically acceptable solvents such as water, ethanol, and the like. Hydrates are formed when the solvent is water, or alcoholates are formed when the solvent is alcohol. Solvates of compounds described herein are conveniently prepared or formed during the processes described herein. In addition, the compounds provided herein optionally exist in unsolvated as well as solvated forms. [0077] In some embodiments, sites on the organic radicals (e.g., alkyl groups, aromatic rings) of compounds of Formula (I), (I-A-8), (I-A-9), or (I-A-10), are susceptible to various metabolic reactions. Incorporation of appropriate substituents on the organic radicals will reduce, minimize or eliminate this metabolic pathway. In specific embodiments, the appropriate substituent to decrease or eliminate the susceptibility of the aromatic ring to metabolic reactions is, by way of example only, a halogen, deuterium, an alkyl group, a haloalkyl group, or a deuteroalkyl group. [0078] In another embodiment, the compounds described herein are labeled isotopically (e.g., with a radioisotope) or by another other means, including, but not limited to, the use of chromophores or fluorescent moieties, bioluminescent labels, or chemiluminescent labels. [0079] Compounds described herein include isotopically-labeled compounds, which are identical to those recited in the various formulae and structures presented herein, but for the fact that one or more atoms are replaced by an atom having an atomic mass or mass number different from the atomic mass or mass number usually found in nature. Examples of isotopes that can be incorporated into the present compounds include isotopes of hydrogen, carbon, nitrogen, oxygen, sulfur, fluorine chlorine, iodine, phosphorus, such as, for example,
2H,
3H,
13C,
14C,
15N,
18O,
17O,
35S,
18F,
36Cl,
123I,
124I,
125I,
131I,
32P and
33P. In one embodiment, isotopically-labeled compounds described herein, for example those into which radioactive isotopes such as
3H and
14C are incorporated, are useful in drug and/or substrate tissue distribution assays. In one embodiment, substitution with isotopes such as deuterium affords certain therapeutic advantages resulting from greater metabolic stability, such as, for example, increased in vivo half-life or reduced dosage requirements. In some embodiments, one or more hydrogens of the compounds of Formula (I), (I-A-8), (I-A- 9), or (I-A-10), are replaced with deuterium. [0080] In some embodiments, a compound represented by the structure of Formula (I), (I-A-8), (I-A-9), or (I-A-10), for example any compound described in Table 1, possesses one or more stereocenters and each stereocenter exists independently in either the R or S configuration. In some embodiments, a compound represented by the structure of Formula (I), (I-A-8), (I-A-9), or (I-A-10), for example any compound described in Table 1, exists in the R configuration. In some embodiments, a compound represented by the structure of Formula (I), (I-A-8), (I-A-9), or (I-A-10), for example any compound described in Table 1, exists in the S configuration. The compounds presented herein include all diastereomeric, individual
WSGR Docket No.55776-726.601 enantiomers, atropisomers, and epimeric forms as well as the appropriate mixtures thereof. The compounds and methods provided herein include all cis, trans, syn, anti, entgegen (E), and zusammen (Z) isomers as well as the appropriate mixtures thereof. [0081] In some embodiments, a composition provided herein comprises a racemic mixture of a compound represented by a structure of Formula (I), (I-A-8), (I-A-9), or (I-A-10), for example any compound described in Table 1. In some embodiments, a compound provided herein is a racemate of a compound represented by a structure of Formula (I), (I-A-8), (I-A-9), or (I-A-10), for example any compound described in Table 1. [0082] Individual stereoisomers are obtained, if desired, by methods such as, stereoselective synthesis and/or the separation of stereoisomers by chiral chromatographic columns or the separation of diastereomers by either non-chiral or chiral chromatographic columns or crystallization and recrystallization in a proper solvent or a mixture of solvents. In certain embodiments, a compound represented by the structure of Formula (I), (I-A-8), (I-A-9), or (I-A-10), for example any compound described in Table 1, is prepared as their individual stereoisomers by reacting a racemic mixture of the compound with an optically active resolving agent to form a pair of diastereoisomeric compounds/salts, separating the diastereomers and recovering the optically pure individual enantiomers. In some embodiments, resolution of individual enantiomers is carried out using covalent diastereomeric derivatives of the compounds described herein. In another embodiment, diastereomers are separated by separation/resolution techniques based upon differences in solubility. In other embodiments, separation of stereoisomers is performed by chromatography or by the forming diastereomeric salts and separation by recrystallization, or chromatography, or any combination thereof. See, e.g., Jean Jacques, Andre Collet, Samuel H. Wilen, “Enantiomers, Racemates and Resolutions”, John Wiley And Sons, Inc., 1981. In some embodiments, stereoisomers are obtained by stereoselective synthesis. [0083] In some embodiments, compounds described herein are prepared as prodrugs. In some embodiments, a prodrug is an agent that is converted into the parent drug in vivo. Prodrugs are often useful because, in some situations, they are easier to administer than the parent drug. In some instances, a prodrug is bioavailable by oral administration whereas the parent is not. Further or alternatively, the prodrug also has improved solubility in pharmaceutical compositions over the parent drug. In some embodiments, the design of a prodrug increases the effective water solubility. An example, without limitation, of a prodrug is a compound described herein, which is administered as an ester (the “prodrug”) but then is metabolically hydrolyzed to provide the active entity. A further example of a prodrug is a short peptide (polyaminoacid) bonded to an acid group where the peptide is metabolized to reveal the active moiety. In certain embodiments, upon in vivo administration, a prodrug is chemically converted to the biologically, pharmaceutically or therapeutically active form of the compound. In certain embodiments, a prodrug is enzymatically metabolized by one or more steps or processes to the biologically, pharmaceutically or therapeutically active form of the compound. [0084] Prodrugs of the compounds described herein include, but are not limited to, esters, ethers, carbonates, thiocarbonates, N-acyl derivatives, N-acyloxyalkyl derivatives, N-alkyloxyacyl derivatives,
WSGR Docket No.55776-726.601 quaternary derivatives of tertiary amines, N-Mannich bases, Schiff bases, amino acid conjugates, phosphate esters, and sulfonate esters. See for example Design of Prodrugs, Bundgaard, A. Ed., Elseview, 1985 and Method in Enzymology, Widder, K. et al., Ed.; Academic, 1985, vol.42, p.309-396; Bundgaard, H. “Design and Application of Prodrugs” in A Textbook of Drug Design and Development, Krosgaard-Larsen and H. Bundgaard, Ed., 1991, Chapter 5, p.113-191; and Bundgaard, H., Advanced Drug Delivery Review, 1992, 8, 1-38, each of which is incorporated herein by reference. In some embodiments, a hydroxyl group in the compounds disclosed herein is used to form a prodrug, wherein the hydroxyl group is incorporated into an acyloxyalkyl ester, alkoxycarbonyloxyalkyl ester, alkyl ester, aryl ester, phosphate ester, sugar ester, ether, and the like. In some embodiments, a hydroxyl group in the compounds disclosed herein is a prodrug wherein the hydroxyl is then metabolized in vivo to provide a carboxylic acid group. In some embodiments, a carboxyl group is used to provide an ester or amide (i.e., the prodrug), which is then metabolized in vivo to provide a carboxylic acid group. In some embodiments, compounds described herein are prepared as alkyl ester prodrugs. [0085] Prodrug forms of the herein described compounds, wherein the prodrug is metabolized in vivo to produce a compound of Formula (I), (I-A-8), (I-A-9), or (I-A-10), as set forth herein are included within the scope of the claims. [0086] In some embodiments, any one of the hydroxyl group(s), amino group(s) and/or carboxylic acid group(s) are functionalized in a suitable manner to provide a prodrug moiety. In some embodiments, the prodrug moiety is as described above. [0087] In additional or further embodiments, the compounds described herein are metabolized upon administration to an organism in need to produce a metabolite that is then used to produce a desired effect, including a desired therapeutic effect. [0088] In some embodiments, a metabolite of a compound disclosed herein is a derivative of that compound that is formed when the compound is metabolized. In some embodiments. an “active metabolite” of a compound provided herein is a biologically active derivative of the compound provided herein that is formed when the compound is metabolized. In some embodiments, metabolism is the sum of the processes (including, but not limited to, hydrolysis reactions and reactions catalyzed by enzymes) by which a particular substance is changed by an organism. In some embodiments, enzymes may produce specific structural alterations to a compound. For example, cytochrome P450 catalyzes a variety of oxidative and reductive reactions while uridine diphosphate glucuronyltransferases catalyze the transfer of an activated glucuronic-acid molecule to aromatic alcohols, aliphatic alcohols, carboxylic acids, amines and free sulfhydryl groups. In some embodiments, a metabolite of a compound disclosed herein is optionally identified either by administration of compounds to a host and analysis of tissue samples from the host, or by incubation of compounds with hepatic cells in vitro and analysis of the resulting compounds.
WSGR Docket No.55776-726.601 Synthesis of Compounds [0089] Compounds of Formula (I), (I-A-8), (I-A-9), or (I-A-10), described herein are synthesized using standard synthetic techniques or using methods known in the art in combination with methods described herein. [0090] Unless otherwise indicated, conventional methods of mass spectroscopy, NMR, HPLC, protein chemistry, biochemistry, recombinant DNA techniques and pharmacology are employed. [0091] Compounds are prepared using standard organic chemistry techniques such as those described in, for example, March’s Advanced Organic Chemistry, 6
th Edition, John Wiley and Sons, Inc. Alternative reaction conditions for the synthetic transformations described herein may be employed such as variation of solvent, reaction temperature, reaction time, as well as different chemical reagents and other reaction conditions. [0092] In some embodiments, compounds described herein are synthesized as outlined in the Examples. Methods of Treatment [0093] In another aspect, disclosed herein is a method of treating a disease or disorder in a subject in need thereof, wherein the method comprises administering to the subject a therapeutically effective amount of the compound of Formula (I), (I-A-8), (I-A-9), or (I-A-10), or a pharmaceutically acceptable salt, pharmaceutically acceptable solvate, diastereomeric mixture, individual enantiomer, or isotopically enriched form thereof. In some embodiments, the disease or disorder is a disease or disorder of the brain. [0094] In some embodiments, the neurological disease or disorder is a neurodegenerative, a neuropsychiatric, or substance use disease or disorder. In some embodiments, the neurological disease or disorder is an injury. In some embodiments, the disease or disorder is an anxiety disorder, a mood disorder, a psychotic disorder, a personality disorder, an eating disorder, a sleep disorder, a sexuality disorder, an impulse control disorder, a substance use disorder, a dissociative disorder, a cognitive disorder, a developmental disorder, or a factitious disorder. In some embodiments, the disease or disorder is a psychotic disorder. In some embodiments, the psychotic disorder is selected from schizophrenia, schizoaffective disorder, schizophreniform disorder, brief psychotic disorder, delusional disorder, shared psychotic disorder, substance-induced psychotic disorder, paraphrenia, psychotic depression, bipolar disorder, schizotypal personality disorder, paranoid personality disorder, schizoid personality disorder, borderline personality disorder, post-traumatic stress disorder, obsessive-compulsive disorder, and dissociative disorders, or psychosis associated with a neurodegenerative disorders. In some embodiments, the neurodegenerative disorder is selected from Huntington’s disease, Alzheimer’s disease, Lewy body dementia, and Parkinson’s disease. In some embodiments, the psychotic disorder is schizophrenia or bipolar disorder. In some embodiments, the method further comprises administering to the subject a therapeutically effective amount of an additional therapeutic agent. [0095] The compounds disclosed herein, or a pharmaceutically acceptable salt, pharmaceutically acceptable solvate, diastereomeric mixture, individual enantiomer, or isotopically enriched form thereof, are useful for promoting neuronal growth and/or improving neuronal structure.
WSGR Docket No.55776-726.601 [0096] In some embodiments, provided herein is a method of promoting neural plasticity (e.g., cortical structural plasticity) in an individual by administering a compound described herein (e.g., a compound represented by the structure of Formula (I), (I-A-8), (I-A-9), or (I-A-10), for example any compound described in Table 1, to the individual. In some embodiments, the individual has or is diagnosed with a brain disorder or other conditions described herein. [0097] In some embodiments, provided herein is a method of promoting neuronal growth in an individual in need thereof, comprising administering to the individual in need thereof a therapeutically effective amount of a compound or pharmaceutical composition provided herein (e.g., a compound having a structure represented by Formula (I), (I-A-8), (I-A-9), or (I-A-10), for example any compound described in Table 1). [0098] In some embodiments, provided herein is a method of improving neuronal structure in an individual in need thereof, comprising administering to the individual in need thereof a therapeutically effective amount of a compound or pharmaceutical composition provided herein (e.g., a compound having a structure represented by Formula (I), (I-A-8), (I-A-9), or (I-A-10), for example any compound described in Table 1). [0099] In some embodiments, provided herein is a method of treating a disease or disorder in an individual in need thereof that is mediated by the loss of synaptic connectivity, plasticity, or a combination thereof, comprising administering to the individual in need thereof a therapeutically effective amount of a compound or pharmaceutical composition provided herein (e.g., a compound having a structure represented by Formula (I), (I-A-8), (I-A-9), or (I-A-10), or for example any compound described in Table 1). [0100] In some embodiments, provided herein is a method of treating a neurological disease or disorder in an individual in need thereof, comprising administering to the individual in need thereof a therapeutically effective amount of a compound or pharmaceutical composition provided herein (e.g., a compound having a structure represented Formula (I), (I-A-8), (I-A-9), or (I-A-10), or for example any compound described in Table 1). [0101] In some embodiments, a compound described herein, or a pharmaceutically acceptable salt thereof, are used in the preparation of medicaments for the treatment of diseases or conditions in a mammal that would benefit from promoting neuronal growth and/or improving neuronal structure. [0102] Methods for treating any of the diseases or conditions described herein in a mammal in need of such treatment, involves administration of pharmaceutical compositions that include at least one compound described herein or a pharmaceutically acceptable salt, active metabolite, prodrug, or pharmaceutically acceptable solvate thereof, in therapeutically effective amounts to said mammal. [0103] In any of the aforementioned aspects are further embodiments in which the effective amount of the compound described herein, or a pharmaceutically acceptable salt thereof, is: (a) systemically administered to the mammal; and/or (b) administered orally to the mammal; and/or (c) intravenously administered to the mammal; and/or (d) administered by injection to the mammal; and/or € administered topically to the mammal; and/or (f) administered non-systemically or locally to the mammal.
WSGR Docket No.55776-726.601 EXAMPLES [0104] The following examples are provided for illustrative purposes only and not to limit the scope of the claims provided herein. General [0105] All reagents are obtained commercially and used without purification unless otherwise noted. DMSO is purified by passage under 12 psi N
2 through activated alumina columns. Reactions are performed using glassware that is flame-dried under reduced pressure (~1 Torr). Compounds purified by chromatography are adsorbed to the silica gel before loading. Thin layer chromatography is performed on Millipore silica gel 60 F254 Silica Gel plates. Visualization of the developed chromatogram is accomplished by fluorescence quenching or by staining with ninhydrin or aqueous ceric ammonium molybdate (CAM). [0106] Nuclear magnetic resonance (NMR) spectra are acquired on either a Bruker 400 operating at 400 and 100 MHz, a Varian 400 operating at 400 and 100 MHz, or a Varian 500 operating at 500 and 125 MHz for
1H and
13C, respectively, and are referenced internally according to residual solvent signals. Data for
1H NMR are recorded as follows: chemical shift (δ, ppm), multiplicity (s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet), coupling constant (Hz), and integration. Data for
13C NMR are reported in terms of chemical shift (δ, ppm). Liquid chromatography-mass spectrometry (LC-MS) is performed using an Agilent LC-MS with Ion Trap or ELSD detector, or a Waters LC-MS with an UPLC detector.
WSGR Docket No.55776-726.601 Chemistry Example A1: [0107] Synthesis of Compound 9, 10, 11, 12, 13, and 14 [0108] Scheme-1: Synthesis of Compound 9, 10, 11, 12, 13, and 14
[0109] Step 1: Synthesis of RP-1 (7-bromo-4,5-difluoro-1H-indole):
WSGR Docket No.55776-726.601 [0110] To a stirred
mmol, 1.0 eq) in THF (190 mL) at -78 °C was added a vinyl magnesium bromide solution (1M in THF, 32.2 g, 319 mmol, 4.0 eq). The reaction mixture was allowed to warm to room temperature and was stirred at that temperature for 12 h. The reaction mixture was quenched with an aqueous NH4Cl solution and extracted with multiple portions of EtOAc. The combined organic extracts were dried over anhydrous Na2SO4, the solids were removed by filtration, and the filtrate was concentrated in vacuo to yield crude product. The crude material was purified by silica gel chromatography to afford 7-bromo-4,5-difluoro-1H-indole (RP-1) (6.2 g, 33%) as a brown liquid. ESI-MS m/z: 231.8 [M-H]
+. [0111] Step 2: Synthesis of RP-2 (1-allyl-7-bromo-4,5-difluoro-1H-indole):
[0112] To a stirred solution of RP-1 (6.2 g, 26.7 mmol, 1.0 eq) in DMF (62 mL) at 0 ℃ was added NaH (60% in mineral oil, 1.28 g, 53.4 mmol, 2.0 eq). The reaction mixture was stirred for 20 min at that temperature and allyl bromide (3.87 g, 32 mmol, 1.2 eq) was added. The reaction mixture was warmed to room temperature and stirred for 3 h. The reaction was diluted with ice cold water and extracted with multiple portions of EtOAc. The combined organic extracts were dried over anhydrous Na2SO4, the solids were removed by filtration, and the filtrate was concentrated in vacuo to yield crude product. The crude material was purified by silica gel chromatography to afford 1-allyl-7-bromo-4,5-difluoro-1H-indole (RP-2) (6.5 g, 90%) as a brown liquid. ESI-MS m/z: 290.1 [M+H2O]
+. [0113] Step 3: RP-
[0114] To a stirred solution of RP-2 (2.0 g, 7.35 mmol, 1.0 eq) in DMF (20 mL) was added K2CO3 (2.0 g, 14.7 mmol, 2.0 eq) at room temperature and the reaction was degassed under N2 atmosphere for 20 minutes. To this reaction mixture was added TBAI (4.06 g, 11 mmol, 1.5 eq) followed by Pd(OAc)2 (0.33 g, 1.47 mmol, 0.2 eq). The reaction mixture was heated at 80 ℃ and was stirred at that temperature for 3 h. The reaction mixture was cooled to room temperature, diluted with water (60 mL), filtered through a pad of
WSGR Docket No.55776-726.601 celite, and the filter pad was washed with EtOAc. The combined organic phase was dried over anhydrous Na
2SO
4, the solids were removed by filtration, and the filtrate was concentrated in vacuo to obtain the crude. The crude was purified by silica gel chromatography column to afford RP-3 (0.75 g, 53%) as a mixture of regio-isomers. ESI-MS m/z:190.2 [M+H]
+. [0115] Step 4: Synthesis of RP-4 (8,9-difluoro-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5-ol):

[0116] To a stirred solution of RP-3 (regio-isomeric mixture, 350 mg, 1.82 mmol, 1.0 eq) in THF (6 mL) at 0 ℃ was added a solution of BH3 (1M in THF, 50.3 mg, 3.64 mmol, 2.0 eq). The reaction mixture was heated to reflux and was stirred at that temperature for 2 h. The reaction mixture was allowed to cool to room temperature, quenched with an aqueous NaOH solution (3M in water, 5 mL) and an aqueous solution of H2O2 (30% in water, 3.5 mL) was added. The reaction mixture was stirred for additional 5 h at room temperature. The reaction mixture was quenched with ice cold aqueous solution of NaCl and extracted with multiple portions of EtOAc. The combined organic layers were dried over anhydrous Na2SO4, the solids were filtered, and the filtrate was concentrated in vacuo to obtain crude material. The crude was purified by silica gel chromatography to afford RP-4 (8,9-difluoro-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5-ol) (220 mg, 58%) as a brown liquid. ESI-MS m/z: 210.0 [M+H]
+. [0117] Step 5: Synthesis of RP-5 (8,9-difluoro-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5-yl 4- methylbenzenesulfonate):
[0118] To a stirred solution of RP-4 (170 mg, 0.81 mmol, 1.0 eq) in dichloromethane (10 mL) was added triethylamine (163 mg, 1.62 mmol, 2.0 eq) at 0 ℃ and the reaction mixture was stirred for 10 mins. To the reaction mixture was added TsCl (229 mg, 1.21 mmol, 1.5 eq), followed by DMAP (9.92 mg, 0.08 mmol, 0.1 eq). The reaction mixture was warmed to room temperature and stirred at that temperature for 6 h. The reaction mixture was diluted with ice cold water and extracted with multiple portions of EtOAc. The combined organic layers were washed with an aqueous solution of NaHCO
3, dried over anhydrous Na
2SO
4, the solids were removed by filtration, and the filtrate was concentrated in vacuo to obtain crude product. The crude material was purified by silica gel chromatography to afford RP-5 (8,9-difluoro-5,6-dihydro-4H- pyrrolo[3,2,1-ij]quinolin-5-yl 4-methylbenzenesulfonate) (110 mg, 37%) as a brown liquid, which was carried forward without further purification.
1H NMR (400 MHz, CHLOROFORM-d) δ: 7.73 (d, J = 7.8
WSGR Docket No.55776-726.601 Hz, 2H), 7.34 (d, J = 7.8 Hz, 2H), 6.98 (d, J = 2.4 Hz, 1H), 6.66 (dd, J = 6.8, 10 Hz, 1H), 6.54 (br s, 1H), 5.35 - 5.11 (m, 1H), 4.29 (d, J = 3.4 Hz, 2H), 3.10 (s, 2H), 2.47 (s, 3H) ppm. [0119] Step 6: Synthesis of RP-6 (5-azido-8,9-difluoro-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinoline):
[0120] To a stirred solution mg, mL) at 0 ℃ was added NaN
3 (28.1 mg, 0.43 mmol, 1.5 eq). The reaction mixture was allowed to warm to room temperature, was heated to 65 ℃, and was stirred at that temperature for 2 h. The reaction mixture was diluted with ice cold water and extracted with multiple portions of diethyl ether. The combined organic layers were washed with ice cold water, followed by an aqueous solution of NaCl. The organic phase was dried over anhydrous Na
2SO
4, the solids were filtered, and the filtrate was concentrated in vacuo to afford crude 5-azido-8,9-difluoro-5,6- dihydro-4H-pyrrolo[3,2,1-ij]quinoline (RP-6) (60 mg, 83%). The crude was directly used in the next step without purification. ESI-MS m/z: 235.3 [M+H]
+. [0121] Step 7: Synthesis of Compound 9 (8,9-difluoro-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5- amine)
[0122] To a stirred solution of crude RP-6 (60 mg, 0.24 mmol, 1.0 eq) in THF (0.6 mL) and H2O (0.3 mL) at 0 ℃ was added TPP (94.6 mg, 0.36 mmol, 1.5 eq). The reaction mixture was allowed to warm to room temperature, was heated to 80 ℃, and was stirred at that temperature for 2 h. The reaction mixture was allowed to cool at room temperature, diluted with ice cold water, and extracted with multiple portions of EtOAc. The combined organic extracts were washed with an aqueous solution of NaCl, dried over anhydrous Na2SO4, the solids were filtered, and the filtrate was concentrated in vacuo to obtain crude material. The crude product was purified by silica gel chromatography to afford 8,9-difluoro-5,6-dihydro- 4H-pyrrolo[3,2,1-ij]quinolin-5-amine (Compound 9) (30 mg, 56 %). ESI-MS m/z: 209.0 [M+H]
+. [0123] Step 8: Synthesis of Compound 12 (8,9-difluoro-N,N-dimethyl-5,6-dihydro-4H-pyrrolo[3,2,1- ij]quinolin-5-amine):
WSGR Docket No.55776-726.601

[0124] To a stirred solution mg, a : mixture (1:1, 1 mL) was added a solution of formaldehyde (37% in water, 3.36 mg, 0.11 mmol, 1.0 eq) at room temperature. The reaction mixture was stirred at that temperature for 30 mins. The reaction mixture was cooled to 0 ℃ and NaCNBH3 (7.03 mg, 0.11 mmol, 1.0 eq) was added portion-wise. The reaction mixture was warmed to room temperature and was stirred at that temperature for 16 h. The volatiles were removed in vacuo, the crude reaction residue was washed with water, and extracted with multiple portions of EtOAc. The combined organic extracts were washed with an aqueous solution of NaCl, the organic layer was dried over anhydrous Na2SO4, the solids were filtered, and the filtrate was concentrated in vacuo. The crude residue was purified by silica gel chromatography to afford Compound 12 (15 mg, 53%).
1H NMR (400 MHz, METHANOL-d4) δ = 7.34 (d, J = 2.9 Hz, 1H), 6.98 (dd, J = 6.8, 10.8 Hz, 1H), 6.60 (d, J = 2.4 Hz, 1H), 4.58 (d, J = 4.4 Hz, 2H), 4.06 (d, J = 4.9 Hz, 1H), 3.51 - 3.40 (m, 2H), 2.93 (s, 6H) ppm. ESI-MS m/z: 237.1 [M+H]
+. [0125] Step 8+9: Synthesis of RP-8 (tert-butyl (8,9-difluoro-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin- 5-yl)carbamate):
[0126] To a stirred solution of Compound 9 (1.1 g, 5.28 mmol, 1.0 eq) in DCM (15 mL) at 0 ℃ was added triethylamine (1.46 mL, 10.5 mmol, 2.0 eq), followed by Boc2O (969 µL, 4.22 mmol, 0.8 eq), and DMAP (cat). The resulting reaction mixture was slowly warmed to room temperature and stirred at that temperature for 16 h. The reaction mixture was diluted with water and extracted with multiple portions of DCM. The combined organic phase was washed with brine, dried over Na2SO4, the solids were removed by filtration, and the filtrate was concentrated in vacuo to afford crude material. The crude compound was purified by silica gel chromatography to afford RP-8 (1.1 g, 68%) as an off white solid.
1H NMR (400 MHz, CHLOROFORM-d) δ: 7.08 (s, 1H), 6.81 (dd, J = 6.8, 10.4 Hz, 1H), 6.56 (d, J = 2.6 Hz, 1H), 4.52 (s, 2H), 4.28 - 4.21 (m, 1H), 4.14 (d, J = 9.6 Hz, 1H), 3.19 (d, J = 16.2 Hz, 1H), 2.92 (d, J = 15.4 Hz, 1H), 1.42 (br s, 9H) ppm. ESI-MS m/z: 309.2 [M+H]+. [0127] 1.1 g of RP-8 was subjected to chiral separation to get 460 mg of RP-9 (tert-butyl (R)-(8,9-difluoro- 5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5-yl)carbamate)& 450 mg of RP-10 (tert-butyl (S)-(8,9-difluoro- 5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5-yl)carbamate).
WSGR Docket No.55776-726.601 Column : Chiralpak-AD-H(250mm, 30mm, 5µ) Mobile Phase A : 0.1% DEA in n- Hexane Mobile Phase B : EtOH Flow rate : 36.0 mL/min [0128] RP-9:
1H NMR (400 MHz, CHLOROFORM-d) δ = 7.08 (d, J = 2.4 Hz, 1H), 6.81 (dd, J = 6.6, 10.4 Hz, 1H), 6.56 (d, J = 2.9 Hz, 1H), 4.59 - 4.48 (m, 1H), 4.27 - 4.21 (m, 1H), 4.18 - 4.10 (m, 1H), 3.19 (d, J = 16.2 Hz, 1H), 2.96 - 2.89 (m, 1H), 1.41 (br s, 9H) ppm. ESI-MS m/z: 308.7 [M+H]+. [0129] RP-10:
1H NMR (400 MHz, CHLOROFORM-d) δ = 7.08 (d, J = 2.4 Hz, 1H), 6.81 (dd, J = 6.4, 10.8 Hz, 1H), 6.56 (d, J = 2.9 Hz, 1H), 4.53 (d, J = 10.2 Hz, 2H), 4.28 - 4.20 (m, 1H), 4.14 (2 d, J = 9.8 Hz, 1H), 3.19 (d, J = 16.2 Hz, 1H), 2.97 - 2.88 (m, 1H), 1.41 (br s, 9H) ppm. ESI-MS m/z: 308.9 [M+H]+. [0130] Step-10: Synthesis of Compound 10 ((R)-8,9-difluoro-5,6-dihydro-4H-pyrrolo[3,2,1- ij]quinolin-5-amine) & Compound 11 ((S)-8,9-difluoro-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5- amine):

[0131] To a stirred solution of RP-9 (350 mg, 1.13 mmol, 1.0 eq) in DCM (15 mL) at 0 ℃ were added 2,6- lutidine (652 µL, 5.64 mmol, 5.0 eq) and TMSOTf (819 µL, 4.52 mmol, 4.0 eq). The reaction mixture slowly warmed to room temperature and was stirred at that temperature for 4 h. The reaction mixture was quenched with a saturated aqueous NaHCO3 solution and extracted with multiple portions of DCM. The combined organic phase was dried over Na2SO4, the solids were filtered, and the filtrate was concentrated in vacuo to yield crude product. The crude material was purified by silica gel chromatography to afford Compound 10 (250 mg, crude).
1H NMR (400 MHz, DMSO-d6) δ: 7.47 (d, J = 2.4 Hz, 1H), 6.97 (dd, J = 6.8, 11.2 Hz, 1H), 6.51 (d, J = 2.9 Hz, 1H), 4.30 (dd, J = 3.4, 12.2 Hz, 1H), 4.09 (br s, 2H), 3.91 (dd, J = 7.2, 12.2 Hz, 1H), 3.66 - 3.59 (m, 1H), 3.10 (d, J = 3.4 Hz, 1H), 2.86 - 2.78 (m, 1H) ppm. ESI-MS m/z: 209.2 [M+H]+.
WSGR Docket No.55776-726.601 [0132] To a stirred solution of RP-10 (450 mg, 1.45 mol, 1.0 eq) in DCM (15 mL) at 0 ℃ were added 2,6- lutidine (837 µL, 7.24 mmol, 5.0 eq) and TMSOTf (1.04 mL, 5.80 mmol, 4.0 eq). The reaction mixture slowly warmed to room temperature and was stirred at that temperature for 4 h. The reaction mixture was quenched with a saturated aqueous NaHCO
3 solution and extracted with multiple portions of DCM. The combined organic phase was dried over Na
2SO
4, the solids were filtered, and the filtrate was concentrated in vacuo to yield crude product. The crude material was purified by silica gel chromatography to afford Compound 11 (240 mg, 80%).
1H NMR (400 MHz, DMSO-d6) δ: 7.42 (d, J = 2.9 Hz, 1H), 6.91 (dd, J = 6.8, 11.2 Hz, 1H), 6.46 (d, J = 2.4 Hz, 1H), 4.24 (dd, J = 3.6, 12.0 Hz, 1H), 4.04 (s, 2H), 3.81 (dd, J = 7.8, 12.2 Hz, 1H), 3.52 - 3.47 (m, 1H), 3.05 (dd, J = 3.4, 16.0 Hz, 1H), 2.76 - 2.70 (m, 1H) ppm. ESI-MS m/z: 209.3 [M+H]+. [0133] Step-11; Synthesis of Compound 13 ((R)-8,9-difluoro-N,N-dimethyl-5,6-dihydro-4H- pyrrolo[3,2,1-ij]quinolin-5-amine) & Compound 14 ((S)-8,9-difluoro-N,N-dimethyl-5,6-dihydro-4H- pyrrolo[3,2,1-ij]quinolin-5-amine)
[0134] To a stirred solution of Compound 10 (200 mg, 960 µmol, 1.0 eq) in THF/MeOH mixture (1:1, 10 vol) was added a solution of formaldehyde (37% in water, 233 mg, 2.88 mmol, 3.0 eq). The reaction mixture was stirred for 30 minutes, then was cooled to 0 ℃, and NaCNBH
3 (120 mg, 1.91 mmol, 2.0 eq) was added. The reaction mixture was warmed to room temperature and was stirred at that temperature for 12 h. The volatiles were evaporated in vacuo, the obtained crude residue was diluted with water (10 mL), and the entire mixture was extracted twice with a 10% MeOH/DCM solution. The combined organic phase was dried over Na
2SO
4, the solids were filtered, and the filtrate was concentrated in vacuo to afford crude compound. The crude material was purified by silica gel chromatography to afford Compound 13 (120 mg, 53%).
[0135] To a stirred solution of Compound 11 (200 mg, 960 µmol, 1.0 eq) in THF/MeOH mixture (1:1, 10 vol) was added a solution of formaldehyde (37% in water, 233 mg, 2.88 mmol, 3.0 eq). The reaction
WSGR Docket No.55776-726.601 mixture was stirred for 30 minutes, then was cooled to 0 ℃, and NaCNBH
3 (120 mg, 1.91 mmol, 2.0 eq) was added. The reaction mixture was warmed to room temperature and was stirred at that temperature for 12 h. The volatiles were evaporated in vacuo, the obtained crude residue was diluted with water (10 mL), and the entire mixture was extracted twice with a 10% MeOH/DCM solution. The combined organic phase was dried over Na
2SO
4, the solids were filtered, and the filtrate was concentrated in vacuo to afford crude compound. The crude material was purified by silica gel chromatography to afford Compound 14 (105 mg, 46%). Example A2: [0136] Scheme-2: Synthesis of Intermediates GG-6a, GG-6b, GG-6c, and GG-6d
[0137] Synthesis of Intermediate Compound GG-6a: tert-butyl (5,6-dihydro-4H-pyrrolo[3,2,1- ij]quinolin-5-yl)carbamate
[0138] Step-1a: Synthesis of GG-1a: methyl 2-((tert-butoxycarbonyl)amino)-3-(1H-indol-7-yl) propanoate):
WSGR Docket No.55776-726.601
[0139] To a stirred solution of 7-bromo-1H-indole (4.0 g, 20.4 mmol, 1.0 eq) in DMF (100 mL) at room temperature were added Compound GG-2, methyl 2-((tert-butoxycarbonyl)amino)-3-iodopropanoate (6.71 g, 20.4 mmol 1.0 eq) and Zn dust (5.33 g, 81.6 mmol, 4.0 eq). The resulting mixture was purged with a nitrogen atmosphere and then Pd(amphos)
2Cl
2 (715 mg, 1.01 mmol, 0.05 eq) was added. The reaction mixture was stirred at ambient temperature for 12 hours. The reaction was diluted with ice-water (50 mL) and extracted with EtOAc (2 x 30 mL). The combined organic layers were washed with an aqueous solution of NaCl (10 mL), the organic layer was dried over anhydrous Na
2SO
4, solids were removed by filtration, and the filtrate was concentrated in vacuo. The crude material was purified by silica gel chromatography (12% EtOAc/Hexane), to afford methyl 2-((tert-butoxycarbonyl)amino)-3-(1H-indol-7-yl) propanoate), Compound GG-3a (3.8 g, 59% yield) as a pale yellow solid. ESI-MS m/z: 318.9 [M-H]-. [0140] Step-2a: Synthesis of Compound GG-4a: tert-butyl (1-hydroxy-3-(1H-indol-7-yl)propan-2- yl)carbamate

[0141] To a stirred solution of Compound GG-3a (3.8 g, 11.9 mmol, 1.0 eq) in a THF: MeOH mixture (10 vol) at 0 ℃ was added NaBH4 (1.80 g, 47.79 mmol, 4.0 eq). The reaction mixture was warmed to room temperature and stirred at that temperature for 1 hour. The reaction mixture was diluted with ice-water (20 mL) and extracted with EtOAc (2 x 10 mL). The combined organic layers were washed with an aqueous solution of NaCl (15 mL), the organic layer was dried over anhydrous Na2SO4, solids were removed by filtration, and the filtrate was concentrated in vacuo. The crude was purified by silica gel chromatography (15% EtOAc/Heptane), to afford tert-butyl (1-hydroxy-3-(1H-indol-7-yl)propan-2-yl)carbamate, Compound GG-4a (2.5 g, 72% yield) as an off white solid. ESI-MS m/z: 291.3 [M+H]
+. [0142] Step-3a: Synthesis of Compound GG-5a: (2-((tert-butoxycarbonyl) amino)-3-(1H-indol-7-yl) propyl methanesulfonate):
WSGR Docket No.55776-726.601
[0143] To a stirred solution of GG-4a (2.5 g, 8.6 mmol, 1.0 eq) in DCM (10 vol) at 0 ℃ was added Et3N (3.65 mL, 25.9 mmol, 3.0 eq), followed by methane sulfonyl chloride (1.32 mL, 17.2 mmol, 2.0 eq). The reaction mixture was warmed to room temperature and stirred at that temperature for 2 hours. An aqueous solution of NaCl (20 mL) was added to quench the reaction and the layers were separated. The organic layer was dried over anhydrous Na2SO4, the solids were removed by filtration, and the filtrate was concentrated in vacuo. Crude (2-((tert-butoxycarbonyl)amino)-3-(1H-indol-7-yl) propyl methanesulfonate), Compound GG- 5a (3 g, crude) was isolated as a pale yellow oil and carried forward without further purification. ESI-MS m/z: 369.2 [M+H]
+. [0144] Step-4a: Synthesis of Compound GG-6a (tert-butyl (5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin- 5-yl) carbamate)):
[0145] To a stirred solution of Compound GG-5a (3 g crude, 8.14 mmol, 1.0 eq) in DMF (10 vol) at 0 ℃ was added NaH (0.78 g, 32.6 mmol, 4.0 eq). The reaction mixture was warmed to room temperature and stirred at that temperature for 2 hours. The reaction mixture was quenched with saturated aqueous NH
4Cl solution (10 mL) and extracted with ethyl acetate (2 x 20 mL). The combined organic layers were dried over anhydrous Na
2SO
4, the solids were removed by filtration, and the filtrate was concentrated in vacuo. The crude material was purified by silica gel chromatography (20% EtOAc/Heptane) to afford (tert-butyl (5,6- dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5-yl) carbamate)), Compound GG-6a (1.3 g, 59% yield) as a white solid. ESI-MS m/z: 273.2 [M+H]
+. [0146] Synthesis of Intermediate GG-6b: tert-butyl (9-chloro-8-fluoro-5,6-dihydro-4H-pyrrolo[3,2,1- ij]quinolin-5-yl)carbamate
WSGR Docket No.55776-726.601
[0147] Step-1b: Synthesis of Int-3b amino)-3-(4-chloro-5-fluor o-1H- indol-7-yl)propanoatebutanoate):

[0148] To a stirred solution of 7-bromo-4-chloro-5-fluoro-1H-indole, Compound GG-1b (3.5 g, 14.1 mmol, 1.0 eq) in DMF (25 vol) were added Compound GG-2 (9.67 g, 28.2 mmol, 2.0 eq) and Zn dust (3.66 g, 56.4 mmol, 4.0 eq) at room temperature. The resulting mixture was purged with a nitrogen atmosphere and Pd(amphos)2Cl2 (0.49 g, 0.7 mmol, 0.05 eq) was added. The reaction mixture was stirred at room temperature for 12 hours. The reaction was diluted with ice-water (30 mL) and extracted with EtOAc (2 x 20 mL). The combined organic layers were washed with an aqueous solution of NaCl (10 mL), the organic layer was dried over anhydrous Na2SO4, solids were removed by filtration, and the filtrate was concentrated in vacuo. The crude material was purified by silica gel chromatography (20% EtOAc/Hexane) to afford (methyl 2-((tert-butoxycarbonyl) amino)-3-(4-chloro-5-fluor o-1H-indol-7-yl)propanoate-butanoate), Compound GG-3b (3.2 g, 62% yield) as a yellow solid. ESI-MS m/z: 369.1 [M-H]-. [0149] Step-2b: Synthesis of Compound GG-4b (tert-butyl (1-(4-chloro-5-fluoro-1H-indol-7-yl)-3- hydroxyp ropan-2-yl) :

[0150] To a stirred solution of Compound GG-3b (3.2 g, 8.64 mmol, 1.0 eq) in MeOH (10 vol) at 0 ℃ was added NaBH
4 (1.30 g, 34.59 mmol, 4.0 eq), portion wise over a period of 20 min. The reaction mixture was warmed to room temperature and stirred at that temperature for 1 hour. The reaction mixture was diluted with ice-water (10 mL) and extracted with EtOAc (2 x 20 mL). The combined organic layers were washed with an aqueous solution of NaCl (15 mL), the organic layer was dried over anhydrous Na
2SO
4, solids were
WSGR Docket No.55776-726.601 removed by filtration, and the filtrate was concentrated in vacuo. The crude material was purified by silica gel chromatography (30% EtOAc/Heptane) to afford (tert-butyl (1-(4-chloro-5-fluoro-1H-indol-7-yl)-3- hydroxypropan-2-yl) carbamate), Compound GG-4b (1.6 g, 54% yield) as an off-white solid. ESI-MS m/z: 342.0 [M+H]
+. [0151] Step-3b: Synthesis of Compound GG-5b: (2-((tert-butoxycarbonyl) amino)-3-(4-chloro-5- fluoro-1H-indol-7-yl) propyl methanesulfonate):
[0152] To a stirred solution of Compound GG-4b (1.6 g, 4.67 mmol, 1.0 eq) in DCM (16 mL) at 0 ℃ was added Et
3N (1.36 mL, 9.35 mmol, 2.0 eq), followed by methane sulfonyl chloride (0.43 mL, 5.60 mmol 1.2 eq). The reaction mixture was warmed to room temperature and stirred at that temperature for 1 hour. The reaction mixture was quenched with saturated aqueous NaHCO
3 solution (10 mL) and extracted with DCM (2 x 10 mL). The combined organic layers were washed with an aqueous solution of NaCl (10 mL) and dried over anhydrous Na
2SO
4. The solids were removed by filtration and the filtrate was concentrated in vacuo to afford crude (2-((tert-butoxycarbonyl) amino)-3-(4-chloro-5-fluoro-1H-indol-7-yl) propyl methanesulfonate), Compound GG-5b (2 g, crude) as a yellow oil, which was carried forward without further purification. ESI-MS m/z: 421.2 [M+H]
+. [0153] Step-4b: Synthesis of Compound GG-6b (tert-butyl (9-chloro-8-fluoro-5, 6-dihydro-4H- pyrrolo[3,2,1-ij]quinolin-5-yl)carbamate):
[0154] To a stirred solution of Compound GG-5b (1.0 g crude, 2.38 mmol, 1.0 eq) in DMF (10 vol) at 0 ℃ was added NaH (228 mg, 9.50 mmol, 4.0 eq) and the reaction mixture was stirred at 0 ℃ for 2 hours. The reaction mixture was quenched with saturated aqueous NH4Cl solution (10 mL) and extracted with ethyl acetate (2 x 20 mL). The combined organic layers were washed with an aqueous solution of NaCl (15 mL), the organic layer was dried over anhydrous Na2SO4, solids were removed by filtration, and the filtrate was concentrated in vacuo. The crude was purified by silica gel chromatography (20% EtOAc/Heptane), to afford (tert-butyl (9-chloro-8-fluoro-5, 6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5-yl)-carbamate), Compound GG-6b (0.3 g, 39% yield) as a yellow solid. ESI-MS m/z: 324.8 [M+H]
+.
WSGR Docket No.55776-726.601 [0155] Synthesis of Intermediate Compound GG-6c: tert-butyl (8-fluoro-5,6-dihydro-4H- pyrrolo[3,2,1-ij]quinolin-5-yl)carbamate
[0156] Step-1c: Synthesis of Compound GG-3c (methyl 2-((tert-butoxycarbonyl)amino)-3-(5-fluoro- 1H-indol-7-yl)propanoate)):

[0157] To a stirred solution of 7-bromo-5-fluoro-1H-indole (3 g, 14.15 mmol, 1.0 eq) in DMF (25 vol) was added Compound GG-2 (9.3 g, 28.3 mmol, 2.0 eq), and Zn (3.66 g, 56.0 mmol, 4.0 eq) at room temperature. The resulting mixture was purged with a nitrogen atmosphere and Pd(amphos)2Cl2 (0.49 g, 700 µmol, 0.05 eq) was added. The reaction mixture was stirred at room temperature for 12 hours. The reaction was quenched with ice-water (50 mL) and extracted with EtOAc (2 x 30 mL). The combined organic layers were washed with an aqueous solution of NaCl (15 mL), the organic layer was dried over anhydrous Na2SO4, the solids were removed by filtration, and the filtrate was concentrated in vacuo. The crude was purified by silica gel chromatography (15% EtOAc/Hexane), to afford methyl 2-((tert-butoxycarbonyl)amino)-3-(5- fluoro-1H-indol-7-yl)propanoate)), Compound GG-3c (2.8 g, 60% yield) as an off-white solid. ESI-MS m/z: 280.7 [M-56] -. [0158] Step-2c: Synthesis of Compound GG-4c: (tert-butyl (1-(5-fluoro-1H-indol-7-yl)-3- hydroxypropan-2-yl)

[0159] To a stirred solution of Compound GG-3c (2.8 g, 8.32 mmol, 1.0 eq) in a THF : MeOH mixture (10 vol) at 0 ℃ was added NaBH
4 (1.25 g, 33.2 mmol, 4.0 eq). The reaction mixture was warmed to room temperature and stirred at that temperature for 1 hour. The reaction mixture was diluted with ice-water (20
WSGR Docket No.55776-726.601 mL) and extracted with EtOAc (2 x 10 mL). The combined organic layers were washed with an aqueous solution of NaCl (15 mL), the organic layer was dried over anhydrous Na
2SO
4, the solids were removed by filtration, and the filtrate was concentrated in vacuo. The crude was purified by silica gel chromatography (15% EtOAc/Heptane) to afford (tert-butyl (1-(5-fluoro-1H-indol-7-yl)-3-hydroxypropan-2-yl) carbamate), Compound GG-4c (2.1 g, 82% yield) as an off white solid. ESI-MS m/z: 309.2 [M+H]
+. [0160] Step: 3c: Synthesis of Compound GG-5c: (2-((tert-butoxycarbonyl) amino)-3-(5-fluoro-1H- indol-7-yl)propylmehanesulfonate):
[0161] To a solution of Compound GG-4c (2.1 g, 6.81 mmol, 1.0 eq), in DCM (10 vol) at 0 ℃ was added Et
3N (2.96 mL, 20.4 mmol, 3.0 eq), followed by methane sulfonyl chloride (11.0 mL, 13.6 mmol, 2.0 eq). Then the reaction mixture was stirred at room temperature for 2 hours. The reaction mixture was quenched with a saturated aqueous NaCl solution (20 mL), the layers were separated, and the organic phase was dried over anhydrous Na
2SO
4. The solids were removed by filtration and the filtrate was concentrated in vacuo, to give (2-((tert-butoxycarbonyl) amino)-3-(5-fluoro-1H-indol-7-yl)propylmehanesulfonate), Compound GG- 5c (2.8 g, crude) as a pale yellow oil, which was carried forward without further purification. ESI-MS m/z: 386.8 [M+H]
+. [0162] Step-4c: Synthesis of Compound GG-6c: tert-butyl (8-fluoro-5,6-dihydro-4H-pyrrolo[3,2,1- ij]quinolin-5-yl)carbamate
[0163] To a solution of Compound GG-5c (2.8 g crude, 7.24 mmol, 1.0 eq) in DMF (28 mL) at 0 ℃ was added NaH (0.69 g, 29.0 mmol, 4.0 eq). The resulting reaction mixture was warmed to room temperature and stirred at that temperature for 2 hours. The reaction mixture was quenched with saturated NH4Cl solution (15 mL) and extracted with ethyl acetate (2 x 20 mL). The combined organic phase was dried over anhydrous Na2SO4, the solids were removed by filtration, and the filtrate was concentrated in vacuo. The crude material was purified by silica gel chromatography (20 % EtOAc/Heptane), to afford tert-butyl (8- fluoro-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5-yl)carbamate, Compound GG-6c (1.4 g, 67% yield) as an off white solid. ESI-MS m/z: 290.9 [M+H]
+.
WSGR Docket No.55776-726.601 [0164] Synthesis of Intermediate Compound GG-6d: tert-butyl (8-chloro-9-fluoro-5,6-dihydro-4H- pyrrolo[3,2,1-ij]quinolin-5-yl)carbamate):
[0165] Step-1d: Synthesis of Compound GG-3d (methyl 2-((tert-butoxycarbonyl)amino)-3-(5-chloro- 4-fluor o-1H-indol-7-yl)propanoate):

[0166] To a solution of 7-bromo-5-chloro-4-fluoro-1H-indole, Compound GG-1d (3.0 g, 12.2 mmol 1.0 eq) in DMF (25 vol) at room temperature were added Compound GG-2 (8.02 g, 24.4 mmol 2.0 eq) and Zn dust (3.17 g, 48.0 mmol, 4.0 eq). The resulting mixture was purged with a nitrogen atmosphere and Pd(amphos)2Cl2 (0.42 g, 0.60 mmol, 0.05 eq) was added. The reaction mixture was stirred at room temperature for 12 hours. The reaction mixture was diluted with ice-water (30 mL) and extracted with EtOAc (2 x 20 mL). The combined organic layers were washed with an aqueous solution of NaCl (10 mL), the organic layer was dried over anhydrous Na2SO4, solids were removed by filtration, and the filtrate was concentrated in vacuo. The crude was purified by silica gel chromatography (20% EtOAc/Hexane) to afford (methyl 2-((tert-butoxycarbonyl)amino)-3-(5-chloro-4-fluor o-1H-indol-7-yl)-propanoate), Compound GG- 3d (3.1 g, 70% yield) as a yellow solid. ESI-MS m/z: 369.1 [M-H]-. [0167] Step-2d: Synthesis of Compound GG-4d: (tert-butyl (1-(5-chloro-4-fluoro-1H-indol-7-yl)-3- hydroxypropan-2-yl)

[0168] To a solution of Compound GG-3d (3.1 g, 8.36 mmol, 1.0 eq) in MeOH (10 vol) at 0 ℃ was added NaBH
4 (1.26 g, 33.4 mmol, 4.0 eq). The resulting reaction mixture was warmed to room temperature and stirred at that temperature for 1 hour. The reaction mixture was diluted with ice-water (10 mL) and extracted
WSGR Docket No.55776-726.601 with EtOAc (2 x 20 mL). The combined organic layers were washed with an aqueous solution of NaCl (15 mL), the organic layer was dried over anhydrous Na
2SO
4, the solids were removed by filtration, and the filtrate was concentrated in vacuo. The crude was purified by silica gel chromatography (30% EtOAc/Heptane), to afford (tert-butyl (1-(5-chloro-4-fluoro-1H-indol-7-yl)-3-hydroxypropan-2- yl)carbamate), Compound GG-4d (1.8 g, 64% yield) as an off-white solid. ESI-MS m/z: 342.9 [M+H]
+. [0169] Step-3d: Synthesis of Compound GG-5d: 2-((tert-butoxycarbonyl)amino)-3-(5-chloro-4-fluoro- 1H-In dol-7-yl)propyl methanesulfonate):
[0170] To a solution of Compound GG-4d (1.6 g, 4.67 mmol, 1.0 eq), in DCM (16 mL) at 0 ℃ was added Et
3N (1.36 mL, 9.32 mmol, 2.0 eq), followed by methanesulfonyl chloride (0.43 mL, 5.60 mmol 1.2 eq). The reaction mixture was warmed to room temperature and stirred at that temperature for 1 hour. The reaction mixture was quenched with a saturated aqueous NaHCO3 solution (20 mL) and extracted with DCM (2 x 20 mL). The combined organic layers were washed with an aqueous solution of NaCl (20 mL) and dried over anhydrous Na
2SO
4. The solids were removed by filtration and the filtrate was concentrated in vacuo to give crude 2-((tert-butoxycarbonyl)amino)-3-(5-chloro-4-fluoro-1H-In dol-7-yl)propyl methanesulfonate), Compound GG-5d (2 g, crude) as a yellow oil, which was carried forward without further purification. ESI- MS m/z: 421.9. [M+H]
+. [0171] Step-4d; Synthesis of Compound GG-6d: tert-butyl (8-chloro-9-fluoro-5,6-dihydro-4H- pyrrolo[3,2,1-ij]quinolin-5-yl)carbamate):

[0172] To a solution of Compound GG-5d (2.0 g crude, 4.76 mmol, 1.0 eq) in DMF (10 mL) at 0 ℃ was added NaH (457 mg, 19.0 mmol, 4 eq). The reaction mixture was warmed to room temperature and stirred at that temperature for 2 hours. The reaction mixture was quenched with a saturated aqueous NH4Cl solution (10 mL) and extracted with ethyl acetate (2 x 20 mL). The combined organic layers were washed with an aqueous solution of NaCl (20 mL) and dried over anhydrous Na2SO4. The solids were removed by filtration and the filtrate was concentrated in vacuo to give crude material. The crude product was purified by silica gel chromatography (15% EtOAc/Heptane) to afford tert-butyl (8-chloro-9-fluoro-5,6-dihydro-4H-
WSGR Docket No.55776-726.601 pyrrolo[3,2,1-ij]quinolin-5-yl)carbamate), Compound GG-6d (0.6 g, 39% yield) as a pale pink solid. ESI- MS m/z: 324.8 [M+H]
+. Example A3: [0173] Synthesis of 8-fluoro-N,9-dimethyl-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5-amine; (Compound 67): [0174] Scheme-3: Synthesis of 8-fluoro-N,9-dimethyl-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5- amine; (Compound 67):
[0175] Step-1: Synthesis of Compound GG-9: (tert-butyl (8-fluoro-9-methyl-5,6-dihydro-4H- pyrrolo[3,2,1-ij]quiolin-5-yl)carbamate):
[0176] To a stirred solution of Compound GG-6b (50 mg, 0.15 mmol, 1.0 eq) in dioxane (5 mL) was added methylboronic acid (18 mg, 0.30 mmol, 2.0 eq) and KOAc (29.4 mg, 0.30 mmol, 2.0 eq) at room temperature. Nitrogen was bubbled through the reaction mixture for 10 minutes, then X-Phos (14.2 mg, 0.03 mmol, 0.2 eq) was added, followed by Pd2(dba)3 (13.74 mg, 0.015 mmol 0.1 eq). Nitrogen was again bubbled through the reaction mixture for 5 minutes, and then the reaction mixture was heated to 80 ℃ and stirred at that temperature for 6 hours. The reaction mixture was cooled to room temperature, quenched with a saturated aqueous NH4Cl solution (5 mL), and extracted with DCM (2 x 5 mL). The combined organic layers were washed with an aqueous solution of NaCl (10 mL), the organic layer was dried over anhydrous Na
2SO
4, the solids were filtered, and the filtrate was concentrated in vacuo. The crude material was purified by silica gel chromatography (18% EtOAc/Heptane) to afford (tert-butyl (8-fluoro-9-methyl-5,6-dihydro- 4H-pyrrolo[3,2,1-ij]quiolin-5-yl)carbamate), Compound GG-9 (30 mg, 64% yield) as a pale yellow film. ESI-MS m/z: 304.8 [M+H]
+. [0177] Step-2: Synthesis of 8-fluoro-N,9-dimethyl-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5-amine; Compound 67
WSGR Docket No.55776-726.601
To a solution of mg, 0 ℃ was added a LiAlH4 solution (2M in THF, 0.19 mL, 0.39 mmol, 4.0 eq). The reaction mixture was heated to reflux and stirred at that temperature for 2 hours. The reaction mixture was cooled to 0 ℃, quenched with a saturated aqueous NH
4Cl solution (5 mL), and extracted with DCM (2 x 5 mL). The combined organic layers were washed with an aqueous solution of NaCl (10 mL), the organic layer was dried over anhydrous Na
2SO
4, the solids were filtered, and the filtrate was concentrated in vacuo. The crude material was submitted for RP prep HPLC purification to afford 8-fluoro-N,9-dimethyl-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5-amine, Compound 67 (15 mg, 70% yield). ESI-MS m/z: 219.1 [M+H]
+.
1H NMR (400 MHz, DMSO-d
6) δ: 7.32 (d, J = 2.9 Hz, 1H), 6.69 (d, J = 10.5 Hz, 1H), 6.39 (d, J = 3.1 Hz, 1H), 4.32 (dd, J = 3.7, 12.0 Hz, 1H), 3.86 - 3.80 (m, 1H), 3.17 - 3.09 (m, 2H), 2.79 - 2.73 (m, 1H), 2.39 (s, 3H), 2.31 (d, J = 1.3 Hz, 3H). Example A4: [0178] Synthesis of 8-fluoro-N,N,9-trimethyl-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5-amine; Compound 73 [0179] Scheme-4: Synthesis of 8-fluoro-N,N,9-trimethyl-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5- amine; Compound 73:
[0180] Step-1: Synthesis of GG-10:(tert-butyl (8-fluoro-9-methyl-5,6-dihydro-4H-pyrrolo[3,2,1-ij] quinolin-5-yl)(methyl)
WSGR Docket No.55776-726.601 [0181] To a solution of Compound GG-9 (80 mg, 0.26 mmol, 1.0 eq) in DMF (10 vol) at 0 ℃ was added NaH (14.4 mg, 3.94 mmol, 1.5 eq) and the reaction mixture was stirred for 10 min. MeI (0.03 mL, 0.52 mmol, 2.0 eq) was added at 0 ℃ and the resulting mixture was warmed to room temperature and stirred at that temperature for 2 hours. The reaction mixture was diluted with ice cold water (5 mL) and extracted with ethyl acetate (2 x 5 mL). The combined organic layers were washed with an aqueous solution of NaCl (10 mL), the organic layer was dried over anhydrous Na
2SO
4, the solids were removed by filtration, and the filtrate was concentrated in vacuo to afford (tert-butyl (8-fluoro-9-methyl-5,6-dihydro-4H-pyrrolo[3,2,1-ij] quinolin-5-yl)(methyl)carbamate), Compound GG-10 (80 mg, crude) as a pale yellow film. The crude material was carried forward without further purification. ESI-MS m/z: 319.30 [M+H]
+. [0182] Step-2: Synthesis of 8-fluoro-N,N,9-trimethyl-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5- amine; Compound 73:
[0183] To a solution of Compound GG-10 (80 mg, 0.25 mmol, 1.0 eq) in DCM (10 vol) at 0 ℃ was added a LiAlH4 solution (2M in THF, 0.50 mL, 1.0 mmol, 4.0 eq). The reaction mixture was heated to reflux and stirred at that temperature for 2 hours. The reaction mixture was cooled to 0 ℃, quenched with a saturated aqueous NH4Cl solution (5 mL), and extracted with DCM (2 x 5 mL). The combined organic layers were washed with an aqueous solution of NaCl (10 mL), the organic layer was dried over anhydrous Na2SO4, the solids were filtered, and the filtrate was concentrated in vacuo. The crude material was purified by RP prep HPLC purification to give 8-fluoro-N,N,9-trimethyl-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5-amine, Compound 73 (30 mg, 52% yield). ESI-MS m/z: 233.15 [M+H]
+.
1H NMR (400 MHz, DMSO-d6) δ: 7.31 - 7.28 (m, 1H), 6.72 - 6.66 (m, 1H), 6.37 (d, J = 3.1 Hz, 1H), 4.34 - 4.25 (m, 1H), 4.02 - 3.95 (m, 1H), 3.05 - 2.87 (m, 3H), 2.31 - 2.29 (m, 9H) ppm. Prep HPLC purification details: COLUMN x-select C18 (250 * 30mm); 5µm Mobile Phase A 10 mM Ammonium bicarbonate in H
2O Mobile Phase B 100% ACN Flow rate 25 mL/min Instrument ID Prep-14 Gradient (Time/%B) 0/10, 3/10, 18/95, 20/95, 22/10
WSGR Docket No.55776-726.601 Example A5: [0184] Synthesis of (R)-9-chloro-8-fluoro-N-methyl-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5-amine (Compound 62) and (S)-9-chloro-8-fluoro-N-methyl-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5-amine (Compound 61) [0185] Scheme-5: Synthesis of (R)-9-chloro-8-fluoro-N-methyl-5,6-dihydro-4H-pyrrolo[3,2,1- ij]quinolin-5-amine (Compound 62) and (S)-9-chloro-8-fluoro-N-methyl-5,6-dihydro-4H- pyrrolo[3,2,1-ij]quinolin-5-amine (Compound 61):
[0186] Step-1: Chiral Resolution of (tert-butyl (9-chloro-8-fluoro-5, 6-dihydro-4H-pyrrolo[3,2,1- ij]quinolin-5-yl)-carbamate), Compound GG-6b: [0187] 250 mg of Compound GG-6b was submitted to chiral resolution (details below) The faster eluting peak was assigned as Compound GG-6b-Peak1 (60 mg, yellow solid, ESI-MS m/z: 304.8 [M+H]
+), and the more slowly eluting peak was assigned as Compound GG-6b-Peak2 (60 mg, yellow solid, ESI-MS m/z: 304.8 [M+H]
+). Chiral HPLC purification details: COLUMN Amylose-2 (30 x 250 mm, 5μm) Mobile Phase A 0.1% Isopropylamine in n- Hexane Mobile Phase B Isopropylamine Eluent A:B 95:05 Flow rate 50 mL/min Diluent Isopropanol + EtOH Detection wavelength 280 nm [0188] Step-2a: Synthesis of (R)-9-chloro-8-fluoro-N-methyl-5,6-dihydro-4H-pyrrolo[3,2,1- ij]quinolin-5-amine;
WSGR Docket No.55776-726.601 [0189] To a solution of Compound GG-6b-Peak1 (60 mg, 0.18 mmol, 1.0 eq) in DCM (10 vol) at 0 ℃ was added a LiAlH
4 solution (2M in THF, 0.70 mL, 1.41 mmol, 4.0 eq). The reaction mixture was heated to reflux and was stirred at that temperature for 2 hours. The reaction mixture was cooled to 0 ℃, quenched with a saturated aqueous NH
4Cl solution (5 mL), and extracted with DCM (2 x 5 mL). The combined organic layers were washed with an aqueous solution of NaCl, the organic layer was dried over anhydrous Na
2SO
4, the solids were filtered, and the filtrate was concentrated in vacuo. The crude material was purified by RP prep HPLC purification to afford (R)-9-chloro-8-fluoro-N-methyl-5,6-dihydro-4H-pyrrolo[3,2,1- ij]quinolin-5-amine, Compound 62 (15 mg, 34% yield). ESI-MS m/z: 239.1 [M+H]
+.
1H NMR (400 MHz, DMSO-d6) δ: 7.48 - 7.47 (m, 1H), 6.95 - 6.91 (m, 1H), 6.42 (d, J = 3.0 Hz, 1H), 4.33 (dd, J = 3.9, 11.9 Hz, 1H), 3.93 - 3.88 (m, 1H), 3.22 - 3.11 (m, 2H), 2.84 - 2.76 (m, 1H), 2.37 (s, 3H). Prep HPLC purification details: COLUMN x-select C18 (250 * 30mm); 5µm Mobile Phase A 10 mM Ammonium bicarbonate in H2O Mobile Phase B 100% ACN Flow rate 25 mL/min Instrument ID Prep-14 Gradient (Time/%B) 0/10, 3/10, 18/95, 20/95, 22/10 [0190] Step-2b: Synthesis of (S)-9-chloro-8-fluoro-N-methyl-5,6-dihydro-4H-pyrrolo[3,2,1- ij]quinolin-5-amine; Compound 61:
[0191] To a solution of Compound GG-6b-Peak2 (60 mg, 0.18 mmol, 1.0 eq) in DCM (10 vol) at 0 ℃ was added a LiAlH
4 solution (2M in THF, 0.70 mL, 1.41 mmol, 4.0 eq). The reaction mixture was heated to reflux and was stirred at that temperature for 2 hours. The reaction mixture was cooled to 0 ℃, quenched with a saturated aqueous NH
4Cl solution (5 mL), and extracted with DCM (2 x 5 mL). The combined organic layers were washed with an aqueous solution of NaCl, the organic layer was dried over anhydrous Na2SO4, the solids were filtered, and the filtrate was concentrated in vacuo. The crude material was purified by RP prep HPLC purification to afford (S)-9-chloro-8-fluoro-N-methyl-5,6-dihydro-4H-pyrrolo[3,2,1- ij]quinolin-5-amine, Compound 61 (15 mg, 34% yield) . ESI-MS m/z: 239.1 [M+H]
+.
1H NMR (400 MHz, DMSO-d6) δ: 7.49 - 7.47 (m, 1H), 6.94 (d, J = 10.1 Hz, 1H), 6.42 (d, J = 3.0 Hz, 1H), 4.33 (dd, J = 3.8, 12.3 Hz, 1H), 3.92 (dd, J = 6.9, 12.3 Hz, 1H), 3.22 - 3.11 (m, 2H), 2.84 - 2.77 (m, 1H), 2.40 - 2.37 (m, 3H).
WSGR Docket No.55776-726.601 Prep HPLC purification details: COLUMN x-select C18 (250 * 30mm); 5 µm Mobile Phase A 10 mM Ammonium bicarbonate in H2O Mobile Phase B 100% ACN Flow rate 25 mL/min Instrument ID Prep-14 Gradient (Time/%B) 0/10, 3/10, 18/95, 20/95, 22/10 Example A6: [0192] Synthesis of 9-chloro-8-fluoro-N,N-dimethyl-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5- amine, Compound 71 [0193] Scheme-6: Synthesis of 9-chloro-8-fluoro-N,N-dimethyl-5,6-dihydro-4H-pyrrolo[3,2,1- ij]quinolin-5-amine, Compound 71
[0194] Step-1: Synthesis of Compound GG-11; (tert-butyl (9-chloro-8-fluoro-5,6-dihydro-4H- pyrrolo[3,2,1-ij] quinolin-5-yl)(methyl)carbamate):
[0195] To a solution of Compound GG-6b (100 mg, 0.31 mmol, 1.0 eq) in DMF (10 vol) at 0 ℃ was added NaH (14.8 mg, 0.61 mmol, 2.0 eq) and the reaction mixture was stirred at 0 ℃ for 10 minutes. MeI (0.04 mL, 0.61 mmol, 2.0 eq) was added at 0 ℃ and the resulting mixture was warmed to room temperature and stirred at that temperature for 30 min. The reaction mixture was diluted with ice cold water (5 mL) and extracted with ethyl acetate (2 x 5 mL). The combined organic layers were washed with an aqueous NaCl solution (5 mL), dried over anhydrous Na
2SO
4, the solids were filtered, and the filtrate was concentrated in vacuo. Crude (tert-butyl (9-chloro-8-fluoro-5,6-dihydro-4H-pyrrolo[3,2,1-ij] quinolin-5-yl)(methyl)-
WSGR Docket No.55776-726.601 carbamate), Compound GG-11 (100 mg, crude) was isolated as a yellow solid and carried forward without further purification. ESI-MS m/z: 338.9 [M+H]
+ [0196] Step-2: Synthesis of 9-chloro-8-fluoro-N,N-dimethyl-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin- 5-amine; Compound 71
[0197] To a solution of Compound GG-11 (100 mg, 0.29 mmol, 1.0 eq) in DCM (10 vol) at 0 ℃ was added a LiAlH4 solution (2M in THF, 0.29 mL, 0.58 mmol, 2.0 eq). The resulting reaction mixture was heated to reflux and was stirred at that temperature for 2 hours. The reaction mixture was cooled to 0 ℃ and quenched with a saturated aqueous NH
4Cl solution (5 mL). The formed solids were filtered off and washed with multiple portions of ethyl acetate. The combined organic extracts were washed with an aqueous solution of NaCl (10 mL), the organic layer was dried over anhydrous Na
2SO
4, the solids were filtered, and the filtrate was concentrated in vacuo. The crude material was purified by silica gel chromatography (3% MeOH/DCM) to afford 9-chloro-8-fluoro-N,N-dimethyl-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5-amine, Compound 71 (50 mg, 68% yield). ESI-MS m/z: 252.9 [M+H]
+.
1H NMR (400 MHz, DMSO-d
6) δ: 7.49 - 7.46 (m, 1H), 6.95 (br d, J = 10.4 Hz, 1H), 6.42 (br d, J = 2.5 Hz, 1H), 4.38 - 4.32 (m, 1H), 4.12 - 4.07 (m, 1H), 3.13 - 2.99 (m, 3H), 2.30 (s, 6H). Example A7: [0198] Synthesis of 9-chloro-8-fluoro-N-methyl-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5-amine; Compound 68:
[0199] To a solution of Compound GG-6b (40 mg, 0.12 mmol, 1.0 eq) in DCM (10 vol) at 0 ℃ was added a LiAlH
4 solution (2M in THF, 0.15 mL, 0.30 mmol, 2.5 eq). The resulting reaction mixture was heated to reflux and was stirred at that temperature for 2 hours. The reaction mixture was cooled to 0 ℃ and quenched with a saturated aqueous NH
4Cl solution (5 mL). The formed solids were filtered off and washed with multiple portions of ethyl acetate. The combined organic extracts were washed with an aqueous solution of
WSGR Docket No.55776-726.601 NaCl (10 mL), the organic layer was dried over anhydrous Na
2SO
4, the solids were filtered, and the filtrate was concentrated in vacuo. The crude was purified by silica gel chromatography (3% MeOH/DCM) to afford 9-chloro-8-fluoro-N-methyl-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5-amine, Compound 68 (20 mg, 70% yield). ESI-MS m/z: 239.3 [M+H]
+.
1H NMR (400 MHz, DMSO-d
6) δ: 7.47 (br d, J = 2.1 Hz, 1H), 6.93 (br d, J = 10.4 Hz, 1H), 6.41 (br d, J = 2.1 Hz, 1H), 4.32 (br dd, J = 2.7, 12.2 Hz, 1H), 3.89 (br dd, J = 7.0, 12.0 Hz, 1H), 3.13 (br d, J = 13.3 Hz, 2H), 2.82 - 2.75 (m, 1H), 2.36 (s, 3H). Example A8: [0200] Synthesis of (R)-9-chloro-8-fluoro-N,N-dimethyl-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5- amine (Compound 65) and (S)-9-chloro-8-fluoro-N,N-dimethyl-5,6-dihydro-4H-pyrrolo[3,2,1- ij]quinolin-5-amine (Compound 64) [0201] Scheme-7: Synthesis of (R)-9-chloro-8-fluoro-N,N-dimethyl-5,6-dihydro-4H-pyrrolo[3,2,1- ij]quinolin-5-amine (Compound 65) and (S)-9-chloro-8-fluoro-N,N-dimethyl-5,6-dihydro-4H- pyrrolo[3,2,1-ij]quinolin-5-amine (Compound 64)
[0202] Step-1a: Synthesis of Compound GG-12; tert-butyl (R)-(9-chloro-8-fluoro-5,6-dihydro-4H- pyrrolo[3,2 ,1-ij]quinolin-5-yl)(methyl)carbamate):

[0203] To a solution of Compound GG-6b-Peak1 (120 mg, 0.36 mmol, 1.0 eq) in DMF (10 vol) at 0 ℃ was added NaH (13.3 mg, 0.55 mmol, 1.5 eq) and the mixture was stirred at 0 ℃ for 10 minutes. MeI (0.04 mL, 0.72 mmol, 2.0 eq) was added at 0 ℃ and the resulting reaction mixture was warmed to room temperature and stirred at that temperature for 2 hours. The reaction mixture was diluted with ice cold water (5 mL) and extracted with ethyl acetate (2 x 5 mL). The combined organic layers were washed with an aqueous solution of NaCl (5 mL), the organic layer was dried over anhydrous Na2SO4, the solids were removed by filtration, and the filtrate was concentrated in vacuo to afford tert-butyl (R)-(9-chloro-8-fluoro- 5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5-yl)(methyl)carbamate), Compound GG-12 (120 mg, crude) as a
WSGR Docket No.55776-726.601 pale yellow film. This material was carried forward without further purification. ESI-MS m/z: 339.0 [M+H]
+. [0204] Step 1b: Synthesis of Compound GG-13: (tert-butyl (S)-(9-chloro-8-fluoro-5,6-dihydro-4H- pyrrolo[3,2,1-ij]quinolin-5-yl)(methyl)carbamate):
[0205] To a solution of Compound GG-6b-Peak2 (130 mg, 0.40 mmol, 1.0 eq) in DMF (10 vol) at 0 ℃ was added NaH (14.4 mg, 0.60 mmol, 1.5 eq) and the mixture was stirred at 0 ℃ for 10 minutes. MeI (0.049 mL, 0.80 mmol, 2.0 eq) was added at 0 ℃ and the resulting reaction mixture was warmed to room temperature and stirred at that temperature for 2 hours. The reaction mixture was diluted with ice cold water (5 mL) and extracted with ethyl acetate (2 x 5 mL). The combined organic layers were washed with an aqueous solution of NaCl (5 mL), the organic layer was dried over anhydrous Na
2SO
4, the solids were removed by filtration, and the filtrate was concentrated in vacuo to afford (tert-butyl (S)-(9-chloro-8-fluoro- 5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5-yl)(methyl)carbamate), Compound GG-13 (130 mg, crude) as a pale yellow film. This material was carried forward without further purification. ESI-MS m/z: 338.9 [M+H]
+. [0206] Step-2a: Synthesis of (R)-9-chloro-8-fluoro-N,N-dimethyl-5,6-dihydro-4H-pyrrolo[3,2,1- ij]quinolin-5-amine; Compound 65
[0207] To a solution of Compound GG-12 (120 mg, 0.35 mmol, 1.0 eq) in DCM (10 vol) at 0 ℃ was added a LiAlH4 solution (2M in THF, 0.71 mL, 1.42 mmol, 4.0 eq). The reaction mixture was heated to reflux and was stirred at that temperature for 2 hours. The reaction mixture was cooled to 0 ℃, quenched with a saturated aqueous NH4Cl solution (10 mL), and extracted with DCM (2 x 10 mL). The combined organic layers were washed with an aqueous solution of NaCl, the organic layer was dried over anhydrous Na2SO4, the solids were filtered, and the filtrate was concentrated in vacuo. The crude material was purified by RP prep HPLC purification to afford (R)-9-chloro-8-fluoro-N,N-dimethyl-5,6-dihydro-4H-pyrrolo[3,2,1-
WSGR Docket No.55776-726.601 ij]quinolin-5-amine, Compound 65 (20 mg, 22% yield). ESI-MS m/z: 253.1 [M+H]
+.
1H NMR (400 MHz, DMSO-d
6) δ: 7.50 - 7.48 (m, 1H), 6.96 (d, J = 10.3 Hz, 1H), 6.42 (d, J = 3.0 Hz, 1H), 4.38 - 4.32 (m, 1H), 4.09 (dd, J = 8.5, 12.3 Hz, 1H), 3.10 - 2.98 (m, 3H), 2.33 - 2.29 (m, 6H). [0208] Step 2b: Synthesis of (S)-9-chloro-8-fluoro-N,N-dimethyl-5,6-dihydro-4H-pyrrolo[3,2,1- ij]quinolin-5-amine (Compound 64)

[0209] To a solution of Compound GG-13 (130 mg, 0.38 mmol, 1.0 eq) in DCM (5 mL) at 0 ℃ was added a LiAlH4 solution (2M in THF, 0.76 mL, 1.53 mmol, 4.0 eq). The reaction mixture was heated to reflux and was stirred at that temperature for 2 hours. The reaction mixture was cooled to 0 ℃, quenched with a saturated aqueous NH4Cl solution (10 mL), and extracted with DCM (2 x 10 mL). The combined organic layers were washed with an aqueous solution of NaCl, the organic layer was dried over anhydrous Na2SO4, the solids were filtered, and the filtrate was concentrated in vacuo. The crude material was purified by RP prep HPLC purification to afford (S)-9-chloro-8-fluoro-N,N-dimethyl-5,6-dihydro-4H-pyrrolo[3,2,1- ij]quinolin-5-amine, Compound 64 (20 mg, 21% yield). ESI-MS m/z: 253.1 [M+H]
+.
1H NMR (400 MHz, DMSO-d6) δ: 7.50 - 7.48 (m, 1H), 6.96 (d, J = 10.3 Hz, 1H), 6.42 (d, J = 3.0 Hz, 1H), 4.38 - 4.32 (m, 1H), 4.12 - 4.06 (m, 1H), 3.12 - 2.98 (m, 3H), 2.31 (s, 6H). Example A9: [0210] Synthesis of 8-chloro-9-fluoro-N,N-dimethyl-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5- amine; Compound 70 [0211] Scheme-8: Synthesis of 8-chloro-9-fluoro-N,N-dimethyl-5,6-dihydro-4H-pyrrolo[3,2,1- ij]quinolin-5-amine; Compound 70
[0212] Step-1: Synthesis of Compound GG-17: tert-butyl (8-chloro-9-fluoro-5,6-dihydro-4H-pyrrolo [3,2,1-ij]quinolin-5-yl)(methyl)carbamate):
WSGR Docket No.55776-726.601
[0213] To a solution of Compound GG-6d (80 mg, 0.24 mmol, 1.0 eq) in DMF (10 vol) at 0 ℃ was added NaH (8.64 mg, 0.36 mmol, 1.5 eq) and the reaction mixture was stirred at that temperature for 10 minutes. MeI (50.7 mg 0.36 mmol 1.5 eq) was added at 0 ℃ and the resulting mixture was warmed to room temperature and stirred at that temperature for 30 minutes. The reaction mixture was diluted with ice cold water (2 mL) and extracted with ethyl acetate (2 x 2 mL). The combined organic layers were washed with an aqueous solution of NaCl (5 mL), dried over anhydrous Na
2SO
4, the solids were removed by filtration, and the filtrate was concentrated in vacuo. The crude reaction residue was triturated with diethyl ether (2 x 10 mL), to provide tert-butyl (8-chloro-9-fluoro-5,6-dihydro-4H-pyrrolo [3,2,1-ij]quinolin-5- yl)(methyl)carbamate), Compound GG-17 (90 mg, crude), as a pale brown oil, which was carried forward without further purification. ESI-MS m/z: 338.9 [M+H]
+. [0214] Step-2: Synthesis of 8-chloro-9-fluoro-N,N-dimethyl-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin- 5-amine; Compound 70:
[0215] To a solution of Compound GG-17 (90 mg, 0.27 mmol, 1.0 eq) in DCM (10 vol) at 0 ℃ was added a LiAlH
4 solution (2M in THF, 0.32 mL, 0.65 mmol, 2.5 eq). The resulting reaction mixture was heated to reflux and was stirred at that temperature for 2 hours. The reaction mixture was cooled to 0 ℃ and quenched with a saturated aqueous NH
4Cl solution (5 mL). The formed solids were filtered off and washed with multiple portions of ethyl acetate. The combined organic extracts were washed with an aqueous solution of NaCl (10 mL), the organic layer was dried over anhydrous Na2SO4, the solids were filtered, and the filtrate was concentrated in vacuo. The crude was purified by silica gel chromatography (3% MeOH/DCM) to afford 8-chloro-9-fluoro-N,N-dimethyl-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5-amine, Compound 70 (30 mg, 45% yield). ESI-MS m/z: 253.2 [M+H]
+.
1H NMR (400 MHz, DMSO-d6) δ: 7.40 (s, 1H), 6.97 (d, J = 6.3 Hz, 1H), 6.46 (d, J = 3.0 Hz, 1H), 4.36 - 4.30 (m, 1H), 4.08 (br dd, J = 8.6, 12.2 Hz, 1H), 3.04 - 2.93 (m, 3H), 2.30 (s, 6H).
WSGR Docket No.55776-726.601 Example A10: [0216] Synthesis of 8-chloro-9-fluoro-N-methyl-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5-amine; Compound 69:

[0217] To a solution of Compound GG-6d (30 mg, 0.09 mmol, 1.0 eq) in DCM (10 vol) at 0 ℃ was added a LiAlH4 (2M in THF, 0.11 mL, 0.23 mmol, 2.5 eq). The resulting reaction mixture was heated to reflux and was stirred at that temperature for 2 hours. The reaction mixture was cooled to 0 ℃ and quenched with a saturated aqueous NH4Cl solution (5 mL). The formed solids were filtered off and washed with multiple portions of ethyl acetate. The combined organic extracts were washed with an aqueous solution of NaCl (10 mL), the organic layer was dried over anhydrous Na2SO4, the solids were filtered, and the filtrate was concentrated in vacuo. The crude was purified by silica gel chromatography (3% MeOH/DCM) to afford Compound 69 (15 mg, 68% yield). ESI-MS m/z: 253.1 [M+H]
+.
1H NMR (400 MHz, DMSO-d6) δ : 7.42 (d, J = 2.9 Hz, 1H), 6.95 (d, J = 6.3 Hz, 1H), 6.46 (d, J = 3.0 Hz, 1H), 4.32 (br dd, J = 2.9, 11.5 Hz, 1H), 3.90 (br dd, J = 7.1, 12.3 Hz, 1H), 3.18 - 3.12 (m, 2H), 2.81 - 2.74 (m, 1H), 2.37 (s, 3H). Example A11: [0218] Synthesis of 8,9-difluoro-N-methyl-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5-amine; Compound 72:
[0219] To a solution of Compound RP-8 (0.5 g, 1.62 mmol 1.0 eq) in DCM (5 mL) at 0 ℃ was added a LiAlH
4 solution (2M in THF, 3.24 mL, 6.49 mmol, 4.0 eq). The reaction mixture was heated to reflux and stirred at that temperature for 2 hours. The reaction mixture was cooled to 0 ℃, quenched with a saturated aqueous NH
4Cl solution (5 mL), and extracted with DCM (2 x 10 mL). The combined organic layers were washed with an aqueous solution of NaCl, the organic layer was dried over anhydrous Na
2SO
4, the solids were filtered, and the filtrate was concentrated in vacuo. The crude material was purified by silica gel
WSGR Docket No.55776-726.601 chromatography (40% EtOAc/Heptane) to afford 8,9-difluoro-N-methyl-5,6-dihydro-4H-pyrrolo[3,2,1- ij]quinolin-5-amine, Compound 72 (280 mg, 77% yield). ESI-MS m/z: 228.2 [M+H]
+.
1H NMR (400 MHz, DMSO-d
6) δ : 7.41 (d, J = 3.0 Hz, 1H), 6.90 (dd, J = 6.9, 11.4 Hz, 1H), 6.47 (d, J = 3.0 Hz, 1H), 4.33 - 4.29 (m, 1H), 3.86 (dd, J = 7.3, 12.1 Hz, 1H), 3.14 - 3.08 (m, 2H), 2.79 - 2.72 (m, 1H), 2.37 (s, 3H). Example A12: [0220] Synthesis of (R)-8,9-difluoro-N-methyl-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5-amine; Compound 63:

[0221] To a solution of Compound RP-9 (150 mg, 0.48 mmol, 1.0 eq) in DCM (10 vol) at 0 ℃ was added a LiAlH4 solution (2M in THF, 0.36 mL, 0.73 mmol 1.5 eq). The resulting reaction mixture was heated to reflux and was stirred at that temperature for 3 hours. The reaction mixture was cooled to 0 ℃ and quenched with a saturated aqueous NH4Cl solution (5 mL). The formed solids were filtered off and washed with multiple portions of ethyl acetate. The combined organic extracts were washed with an aqueous solution of NaCl (10 mL), the organic layer was dried over anhydrous Na2SO4, the solids were filtered, and the filtrate was concentrated in vacuo. The crude was purified by silica gel chromatography (10% MeOH/DCM) to afford (R)-8,9-difluoro-N-methyl-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5-amine, Compound 63 (100 mg, 90% yield). ESI-MS m/z: 223.1 [M+H]
+.
1H NMR (400 MHz, METHANOL-d4) δ: 7.23 (d, J = 3.1 Hz, 1H), 6.80 (dd, J = 6.7 Hz, 11.2 Hz, 1H), 6.47 (d, J = 3.0 Hz, 1H), 4.36 (ddd, J = 0.9 Hz, 3.9 Hz, 12.1 Hz, 1H), 3.95 (dd, J = 7.4 Hz, 12.3 Hz, 1H), 3.28 - 3.18 (m, 2H), 2.85 (dd, J = 7.8 Hz, 15.8 Hz, 1H), 2.49 (s, 3H). Example A13: [0222] Synthesis of (S)-8,9-difluoro-N-methyl-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5-amine; Compound 66:
WSGR Docket No.55776-726.601 [0223] To a solution of Compound RP-10 (250 mg, 0.810 mmol 1.0 eq) in DCM (10 vol) at 0 ℃ was added a LiAlH
4 solution (2M in THF, 1.21 mL, 2.43 mmol, 3.0 eq). The resulting reaction mixture was heated to reflux and was stirred at that temperature for 3 hours. The reaction mixture was cooled to 0 ℃ and quenched with a saturated aqueous NH
4Cl solution (6 mL). The formed solids were filtered off and washed with multiple portions of ethyl acetate (2 x 6 mL). The combined organic extracts were washed with an aqueous solution of NaCl (10 mL), the organic layer was dried over anhydrous Na
2SO
4, the solids were filtered, and the filtrate was concentrated in vacuo. The crude was purified by silica gel chromatography (10% MeOH/DCM) to afford (S)-8,9-difluoro-N-methyl-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5- amine, Compound 66 (130 mg, 71% yield). ESI-MS m/z: 223.1 [M+H]
+.
1H NMR (400 MHz, METHANOL-d4) δ : 7.24 - 7.22 (m, 1H), 6.83 - 6.77 (m, 1H), 6.47 (d, J = 3.0 Hz, 1H), 4.36 (ddd, J = 0.9 Hz, 3.9 Hz, 12.1 Hz, 1H), 3.97 - 3.92 (m, 1H), 3.29 - 3.20 (m, 2H), 2.85 (dd, J = 7.8 Hz, 15.8 Hz, 1H), 2.49 (s, 3H). Biological Examples Example B-1: Neurite outgrowth assay. [0224] Changes in the pattern of neurite outgrowth have been implicated in neurodegenerative disorders as well as traumatic injuries. The discovery of compounds that can positively affect neuritogenesis are important for developing new therapeutics for neurological diseases. In some instances, measurement of neurite outgrowth of rat cortical neurons using an automated image-based assay is used to determine the neuroplastic effects of the compounds provided herein. In some embodiments, a compound provided herein increases the pattern of neurite outgrowth. In some embodiments, a compound provided herein increases neurite average length compared to a control. In some embodiments, a compound provided herein increases neurite branch points compared to a control. In some embodiments, a compound provided herein increases neurite average length and neurite branch points compared to a control. [0225] In some embodiments, the plastogenic potential of compounds provided herein is assessed by measuring the changes in neurite development. Example B-2: Dendritogenesis Assays. [0226] Phenotypic screening has historically proven more successful than target-based approaches for identifying drugs with novel mechanisms of action. Using a phenotypic assay, the compounds provided herein are tested for their ability to increase dendritic arbor complexity in cultures of cortical neurons. Following treatment, neurons are fixed and visualized using an antibody against MAP2—a cytoskeletal protein localized to the somatodendritic compartment of neurons. Sholl analysis is then performed, and the maximum number of crossings (Nmax) is used as a quantitative metric of dendritic arbor complexity. For statistical comparisons between specific compounds, the raw N
max values are compared. Percent efficacies are determined by setting the N
max values for the vehicle (DMSO) and positive (ketamine) controls equal to 0% and 100%, respectively.
WSGR Docket No.55776-726.601 [0227] Animals. For the dendritogenesis experiments, timed pregnant Sprague Dawley rats are obtained from Charles River Laboratories (Wilmington, MA). In some instances, male and female C57BL/6J mice are obtained from Jackson Laboratory (Sacramento, C.A.). In some instances, mice are housed in a temperature and humidity-controlled room maintained on a 12-h light/dark cycle in groups of 4–5 (same sex). Example B-3: Dendritogenesis – Sholl Analysis. [0228] Neurons are plated in 96-well format (200 ^L of media per well) at a density of approximately 15,000 cells/well in Neurobasal (Life Technologies) containing 1% penicillin-streptomycin, 10% heat- inactivated fetal bovine serum, and 0.5 mM glutamine. After 24 h, the medium is replaced with Neurobasal containing 1x B27 supplement (Life Technologies), 1% penicillin-streptomycin, 0.5 mM glutamine, and 12.5 ^M glutamate. After 3 days in vitro (DIV3), the cells are treated with compounds. Compounds tested in the dendritogenesis assays are treated at 10 ^M unless noted otherwise. Stock solutions of the compounds in DMSO are first diluted 100-fold in Neurobasal before an additional 10-fold dilution into each well (total dilution = 1:1000; 0.1% DMSO concentration). Treatments are randomized. After 1 h, the media is removed and replaced with new Neurobasal media containing 1x B27 supplement, 1% penicillin-streptomycin, 0.5 mM glutamine, and 12.5 ^M glutamate. The cells grow for an additional 71 h. At that time, neurons are fixed by removing 80% of the media and replacing it with a volume of 4% aqueous paraformaldehyde (Alfa Aesar) equal to 50% of the working volume of the well. Then, the cells are incubated at room temperature for 20 min before the fixative is aspirated and each well washed twice with DPBS. Cells are permeabilized using 0.2% Triton X-100 (ThermoFisher) in DPBS for 20 minutes at room temperature without shaking. Plates are blocked with antibody diluting buffer (ADB) containing 2% bovine serum albumin (BSA) in DPBS for 1 h at room temperature. Then, plates are incubated overnight at 4ºC with gentle shaking in ADB containing a chicken anti-MAP2 antibody (1:10,000; EnCor, CPCA-MAP2). The next day, plates are washed three times with DPBS and once with 2% ADB in DPBS. Plates are incubated for 1 h at room temperature in ADB containing an anti-chicken IgG secondary antibody conjugated to Alexa Fluor 488 (Life Technologies, 1:500) and washed five times with DPBS. After the final wash, 100 µL of DPBS is added per well and imaged on an ImageXpress Micro XL High-Content Screening System (Molecular Devices, Sunnyvale,CA) with a 20x objective. [0229] Images are analyzed using ImageJ Fiji (version 1.51W). First, images corresponding to each treatment are sorted into individual folders that are then blinded for data analysis. Plate controls (both positive and negative) are used to ensure that the assay is working properly as well as to visually determine appropriate numerical values for brightness/contrast and thresholding to be applied universally to the remainder of the randomized images. Next, the brightness/contrast settings are applied, and approximately 1–2 individual pyramidal-like neurons per image (i.e., no bipolar neurons) are selected using the rectangular selection tool and saved as separate files. Neurons are selected that did not overlap extensively with other cells or extend far beyond the field of view. The threshold settings are then applied to the individual images. The paintbrush tool is used to eliminate artifacts and dendritic processes originating from adjacent neurons
WSGR Docket No.55776-726.601 (cleanup phaseNext, the point tool is used to select the center of the neuron, and the images are saved and processed using the following Sholl analysis batch macro: run("Sholl Analysis...", "starting=0 ending=NaN radius_step=2 #_samples=1 integration=Mean enclosing=1 #_primary=4 infer fit linear polynomial=[Best fitting degree] most semi-log normalizer=Area create background=228 save do"); [0230] Sholl analysis circle radii = 2 pixel increments = 0.67 ^m. All images are taken and analyzed by an experimenter blinded to treatment conditions. The number of crossings for each neuron at each distinct radius is averaged to produce an average Sholl plot for each treatment. The N
max values are simply
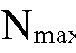
determined by identifying the maximum of each plot. For each treatment, are selected from at least 6 wells spread across 2 plates (9 sites/well x 3 wells/plate x 2 plates). Each plate is prepared using neurons obtained from independent pregnant dams). Example B-4: Spinogenesis Experiments. [0231] Spinogenesis experiments are performed as previously described with the exception that cells are treated on DIV19 and fixed 24 h after treatment on DIV20. (Ly, C. et al., 2018) The images are taken on a Nikon HCA Confocal microscope a with a 100x/NA 1.45 oil objective. DMSO and ketamine (10 ^M) are used as vehicle and positive controls, respectively. Example B-5: Serotonin 5-HT2C In Vitro Cellular IPOne Agonism Assay. [0232] The 5-HT2C IPOne HTRF assay was performed at Epics Therapeutics S.A. (Belgium, FAST-0507I) using conventional methods. Briefly, CHO-K1 cells expressing human recombinant 5-HT2Cedited receptor (accession number AAF35842.1) grown to mid-log phase in culture media without antibiotics were detached with PBS-EDTA, centrifuged, and resuspended in medium without antibiotics buffer.20,000 cells are distributed in a 96 well plate and incubated overnight at 37°C with 5% CO
2. [0233] For agonist testing, the medium was removed and 20μl of assay buffer plus 20μl of test compound or reference agonist are added in each well. The plate is incubated for 60 min. at 37°C with 5% CO
2. [0234] After addition of the lysis buffer containing IP1-d2 and anti-IP1 cryptate detection reagents, plates were incubated 1-hour at room temperature, and fluorescence ratios were measured according to the manufacturer specification, with the HTRF kit. [0235] The compounds provided herein were tested in the Serotonin 5-HT2A and 5-HT2C in vitro radioligand binding and cellular IPOne agonism assays. The binding and agonism functional potencies of the compounds (as indicated by their IC50s or EC50s) are shown in Table 7. Example B-6: Serotonin 5-HT2A In Vitro Cellular IPOne Antagonism Assay. [0236] The 5-HT2A IPOne HTRF assay was performed at Epics Therapeutics S.A. (Belgium, FAST-0505I) in antagonism mode using conventional methods. Briefly, CHO-K1 cells expressing human recombinant 5- HT2A or 5-HT2C receptor grown to mid-log phase in culture media without antibiotics were detached with PBS-EDTA, centrifuged, and resuspended in medium without antibiotics buffer.20,000 cells were distributed in a 96 well plate and incubated overnight at 37°C with 5% CO2.
WSGR Docket No.55776-726.601 [0237] For antagonist testing, a reference agonist a-Me-5HT was added and fluorescence signal monitored for several minutes, followed by addition of 20μl of assay buffer plus 20μl of test compound or reference antagonist ketanserin, in each well. The plate was incubated for 60 min. at 37°C with 5% CO
2. [0238] After addition of the lysis buffer containing IP1-d2 and anti-IP1 cryptate detection reagents, plates were incubated 1-hour at room temperature, and fluorescence ratios were measured according to the manufacturer specification, with the HTRF kit. [0239] The compounds provided herein were tested in the Serotonin 5-HT2A antagonist and Serotonin 5- HT2C cellular IPOne agonist assay. The agonist and antagonist functional potencies of the compounds are shown in Table 2. Table 2 5HT2A IPOne 5HT2C IPOne Compound Number Antagonism Activity Agonism Activity 12 B B 13 C C 14 B B 61 B B 6
2 D C 6
3 E C 6
4 D C 6
5 C B 6
6 D C 6
7 C B 6
8 C B 6
9 C B 7
0 E C 7
1 D C 7
2 C B 7
3 C B Comparator C
C A: IC50 or EC50 is <0.010 ^M; B: IC50 or EC50 is 0.010 ^M - 0.100 ^M; C: IC50 or EC50 is 0.101 ^M - 1 ^M; D: IC50 or EC50 is 1.001 ^M - 10 ^M; and E: IC50 or EC50 is >10 ^M
WSGR Docket No.55776-726.601 Example B-7: Neurite Outgrowth Assay in Primary Neuronal Cultures (Cellectricon). [0240] Changes in the pattern of neurite outgrowth have been implicated in psychiatric and neurodegenerative disorders as well as traumatic injuries. The discovery of new compounds that can positively affect neuritogenesis are important for developing new therapeutics for neurological diseases. Measurement of neurite outgrowth of rat cortical neurons using an automated image-based assay was used to determine the neuroplastic effects of the compounds of the present disclosure. The neurite outgrowth assay was performed at Cellectricon AB (Sweden) as described below. [0241] All experimental work was conducted in accordance with European and Swedish animal welfare regulations. Pregnant Wistar Han rats (Janvier Labs, France) were housed and the cortices microsurgically dissected at University of Gothenburg, dept. Experimental Biomedicine by trained Cellectricon staff at gestation day 17 ½. The cortical cell culture preparations were performed in the cell laboratory at Cellectricon under sterile conditions. [0242] Cortices were collected in microtubes with Hibernate E on ice. The tissue was triturated with a sterile glass Pasteur pipette to dissociate the tissue and the supernatant from each tube was transferred and pooled into one tube. To each remaining pellet, fresh Hibernate E was added. The trituration procedure above was repeated to fully dissociate the tissue. After the final trituration, the cell suspension was centrifuged. The supernatant was removed, and the pellet re-suspended by addition of Neurobasal complete medium. The cell suspensions were triturated between each addition to dissociate cell aggregates. The cells were counted using an automated cell counter (Countess II 2.0, Invitrogen). [0243] Cells were added to wells in Cellaxess Elektra Poly-D-Lysine coated 384-well plates (Cellectricon AB) at a density of 2500 cells/well. Cells were cultured at 37°C, 5% CO2, 95% humidity. [0244] Test compounds were added after 3 days in vitro (DIV) to the cell cultures in concentration response format. The reference compound DL-2,5-Dimethoxy-4-iodoamphetamine hydrochloride (DOI, #10998.11, Chiron/Lab Sweden) was added in a fixed concentration (10 µM) to each plate. [0245] At 9 DIV, cell cultures were fixated using 4% PFA. After fixation, plates were blocked with a PBS- based solution consisting of 2% normal goat serum, 0.2% Triton X-100 and stained with primary antibody (anti-β-tubulin III; 1:1000) for detecting the axonal network, incubated overnight and thereafter with secondary antibody. In addition, cultures were stained with Hoechst (1:10000) for nuclear staining. [0246] High content data was acquired using a Perkin Elmer Operetta high content imager. Data analysis was carried out using the Perkin Elmer Harmony analysis software. To evaluate total number of cells, nuclei were identified using a predefined nuclear detection step. For quantification of total neurite length, the anti- β-III-tubulin positive neurites were detected with a predefined detection step, with parameters adjusted to aid optimal detection of the neurite network staining. [0247] For quantification of parameters on a single cell level, neurites were allocated to individual cell bodies. The cell body of an individual neuron was determined as the cytoplasm around the previously selected nucleus in a median smoothed image of the anti-β-III-tubulin staining.
WSGR Docket No.55776-726.601 [0248] Compound effect on neurite outgrowth was evaluated based on two parameters: “number of type one nodes” (single cell level) and “total segment length” (population level). Total segment length data was fitted to Hill’s equation and EC
50 values were determined for active compounds. [0249] Plastogenic potential (as measured by Neurite Outgrowth Assay) of compounds disclosed herein are shown in Table 3. Table 3 Compound Increase in Neurite Number Length 12 A 13 A 14 A 6
1 A 6
2 A 6
3 B 6
4 A 6
5 B 6
6 A 6
7 B 6
8 B 6
9 A 7
0 A 7
1 A 7
3 A Comparator B A: Statistically significant mean increase as a percent of DMSO control at two adjacent concentrations of 10 uM or less B: No statistically significant mean increase as a percent of DMSO control at two adjacent concentrations of 10 uM or less [0250] Compounds in Example B-6 and B-7 were tested alongside comparator compound (R)-N,N- dimethyl-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5-amine. As shown in Table 3, the comparator compound is unable to increase neurite length at concentrations up to 10 uM, while compounds described herein are potent neuroplastogens. [0251] The examples and embodiments described herein are for illustrative purposes only and various modifications or changes suggested to persons skilled in the art are to be included within the spirit and purview of this application and scope of the appended claims.
 Formula (I-A-8). [0048] In some embodiments, the compound of Formula (I) has the structure of Formula (I-A-9), or a pharmaceutically acceptable salt, pharmaceutically acceptable solvate, diastereomeric mixture, individual enantiomer, or isotopically enriched form thereof:
Formula (I-A-8). [0048] In some embodiments, the compound of Formula (I) has the structure of Formula (I-A-9), or a pharmaceutically acceptable salt, pharmaceutically acceptable solvate, diastereomeric mixture, individual enantiomer, or isotopically enriched form thereof:
 Formula (I-A-9). [0049] In some embodiments, the compound of Formula (I) has the structure of Formula (I-A-10), or a pharmaceutically acceptable salt, pharmaceutically acceptable solvate, diastereomeric mixture, individual enantiomer, or isotopically enriched form thereof:
Formula (I-A-9). [0049] In some embodiments, the compound of Formula (I) has the structure of Formula (I-A-10), or a pharmaceutically acceptable salt, pharmaceutically acceptable solvate, diastereomeric mixture, individual enantiomer, or isotopically enriched form thereof:
 Formula (I-A-10). [0050] In some embodiments of Formula (I), (I-A-8), (I-A-9), or (I-A-10), each R3, R4, and R5, when present, is independently hydrogen, C1-C6alkyl, C1-C6haloalkyl, C1-C6hydroxyalkyl, C1-C6aminoalkyl, C1- C6heteroalkyl, halogen, -CN, -NO2, -ORa, -SRa, -NRcRd, -S(=O)Rb, -S(=O)2Rb, -S(=O)2NRcRd, - NRbS(=O)2Rb, -NRbS(=O)2NRcRd, -C(=O)Rb, -C(=O)ORb, -OC(=O)Rb, -OC(=O)ORb, -OC(=O)NRcRd, - NRbC(=O)Rb, -NRbC(=O)ORb, -NRbC(=O)NRcRd, -C(=O)NRcRd, -P(=O)(ORc)(ORd), -P(=O)RcRd, C6-
WSGR Docket No.55776-726.601 C10aryl, 5-10 membered heteroaryl, C3-C7cycloalkyl, 3- to 10- membered heterocycloalkyl, C1-C6alkyl(C3- C7cycloalkyl), C1-C6alkyl(3- to 10- membered heterocycloalkyl), C1-C6alkyl(C6-C10aryl), or C1-C6alkyl(heteroaryl), wherein each of the alkyl, heteroalkyl, aryl, heteroaryl, cycloalkyl, and heterocycloalkyl is optionally substituted with one or more substituents selected from C1-C3alkyl, C1- C3haloalkyl, C1-C3alkoxy, halogen, -OH, -CN, and =O. In some embodiments, each R3, R4, and R5 is independently hydrogen, C1-C6alkyl, C1-C6heteroalkyl, halogen, C6-C10aryl, or C3-C7cycloalkyl, wherein each of the alkyl, heteroalkyl, aryl, and cycloalkyl is optionally substituted with one or more substituents selected from halogen, C1-C3alkyl, C1-C3alkoxy, halogen, -OH, and -CN. In some embodiments, each R3, R4, and R5 is independently hydrogen, C1-C6alkyl, C1-C6heteroalkyl, -F, -Cl, -Br, phenyl, cyclopropyl, or cyclobutyl, wherein each of the alkyl, heteroalkyl, phenyl, cyclopropyl, and cyclobutyl is optionally substituted with one or more substituents selected from C1-C3alkyl, C1-C3alkoxy, -F, -Cl, -Br, and -CN. In some embodiments, each R3, R4, and R5 is independently hydrogen, C1-C6alkyl, C1-C6heteroalkyl, -F, -Cl, - Br, phenyl, cyclopropyl, or cyclobutyl, wherein each of the alkyl, heteroalkyl, phenyl, cyclopropyl, and cyclobutyl is optionally substituted with one or more substituents selected from methyl, -O-CH3, -F, -Cl, -Br, and -CN. In some embodiments, each R3, R4, and R5 is independently hydrogen, -CH3, -F, -Cl, or -Br
Formula (I-A-10). [0050] In some embodiments of Formula (I), (I-A-8), (I-A-9), or (I-A-10), each R3, R4, and R5, when present, is independently hydrogen, C1-C6alkyl, C1-C6haloalkyl, C1-C6hydroxyalkyl, C1-C6aminoalkyl, C1- C6heteroalkyl, halogen, -CN, -NO2, -ORa, -SRa, -NRcRd, -S(=O)Rb, -S(=O)2Rb, -S(=O)2NRcRd, - NRbS(=O)2Rb, -NRbS(=O)2NRcRd, -C(=O)Rb, -C(=O)ORb, -OC(=O)Rb, -OC(=O)ORb, -OC(=O)NRcRd, - NRbC(=O)Rb, -NRbC(=O)ORb, -NRbC(=O)NRcRd, -C(=O)NRcRd, -P(=O)(ORc)(ORd), -P(=O)RcRd, C6-
WSGR Docket No.55776-726.601 C10aryl, 5-10 membered heteroaryl, C3-C7cycloalkyl, 3- to 10- membered heterocycloalkyl, C1-C6alkyl(C3- C7cycloalkyl), C1-C6alkyl(3- to 10- membered heterocycloalkyl), C1-C6alkyl(C6-C10aryl), or C1-C6alkyl(heteroaryl), wherein each of the alkyl, heteroalkyl, aryl, heteroaryl, cycloalkyl, and heterocycloalkyl is optionally substituted with one or more substituents selected from C1-C3alkyl, C1- C3haloalkyl, C1-C3alkoxy, halogen, -OH, -CN, and =O. In some embodiments, each R3, R4, and R5 is independently hydrogen, C1-C6alkyl, C1-C6heteroalkyl, halogen, C6-C10aryl, or C3-C7cycloalkyl, wherein each of the alkyl, heteroalkyl, aryl, and cycloalkyl is optionally substituted with one or more substituents selected from halogen, C1-C3alkyl, C1-C3alkoxy, halogen, -OH, and -CN. In some embodiments, each R3, R4, and R5 is independently hydrogen, C1-C6alkyl, C1-C6heteroalkyl, -F, -Cl, -Br, phenyl, cyclopropyl, or cyclobutyl, wherein each of the alkyl, heteroalkyl, phenyl, cyclopropyl, and cyclobutyl is optionally substituted with one or more substituents selected from C1-C3alkyl, C1-C3alkoxy, -F, -Cl, -Br, and -CN. In some embodiments, each R3, R4, and R5 is independently hydrogen, C1-C6alkyl, C1-C6heteroalkyl, -F, -Cl, - Br, phenyl, cyclopropyl, or cyclobutyl, wherein each of the alkyl, heteroalkyl, phenyl, cyclopropyl, and cyclobutyl is optionally substituted with one or more substituents selected from methyl, -O-CH3, -F, -Cl, -Br, and -CN. In some embodiments, each R3, R4, and R5 is independently hydrogen, -CH3, -F, -Cl, or -Br
 [0051] In some embodiments of Formula (I), (I-A-2), (I-A-5), (I-A-8), (I-A-9), (I-A-11), (I-A-14), (I-A-15), (I-A-17), (I-A-18), or (I-A-20), R3 is hydrogen, C1-C6alkyl, C1-C6heteroalkyl, -F, -Cl, -Br, or phenyl, wherein each of the alkyl, heteroalkyl, and phenyl is optionally substituted with one or more substituents selected from C1-C3alkyl, C1-C3alkoxy, -F, -Cl, -Br, and -CN. In some embodiments, R3 is hydrogen. In some embodiments, R3 is C1-C6alkyl, C1-C6heteroalkyl, -F, -Cl, -Br, or phenyl, wherein each of the alkyl, heteroalkyl, and phenyl is optionally substituted with one or more substituents selected from C1-C3alkyl, C1- C3alkoxy, -F, -Cl, -Br, and -CN. In some embodiments, R3 is C1-C6alkyl, C1-C6heteroalkyl, -F, -Cl, -Br, or phenyl, wherein each of the alkyl, heteroalkyl, and phenyl is optionally substituted with one or more substituents selected from methyl, -O-CH3, -F, -Cl, -Br, and -CN. [0052] In some embodiments of Formula (I), (I-A-3), (I-A-6), (I-A-8), (I-A-10), (I-A-12), (I-A-14), (I-A- 16), (I-A-17), (I-A-19), or (I-A-20), R4 is hydrogen, C1-C6alkyl, C1-C6heteroalkyl, -F, -Cl, -Br, or phenyl, wherein each of the alkyl, heteroalkyl, and phenyl is optionally substituted with one or more substituents selected from C1-C3alkyl, C1-C3alkoxy, -F, -Cl, -Br, and -CN. In some embodiments, R4 is hydrogen. In some embodiments, R4 is C1-C6alkyl, C1-C6heteroalkyl, -F, -Cl, -Br, or phenyl, wherein each of the alkyl, heteroalkyl, and phenyl is optionally substituted with one or more substituents selected from C1-C3alkyl, C1- C3alkoxy, -F, -Cl, -Br, and -CN. In some embodiments, R4 is C1-C6alkyl, C1-C6heteroalkyl, -F, -Cl, -Br, or phenyl, wherein each of the alkyl, heteroalkyl, and phenyl is optionally substituted with one or more substituents selected from methyl, -O-CH3, -F, -Cl, -Br, and -CN.
WSGR Docket No.55776-726.601 [0053] In some embodiments of Formula (I), (I-A-4), (I-A-7), (I-A-9), (I-A-10), (I-A-13), (I-A-15), (I-A- 16), (I-A-18), (I-A-19), or (I-A-20), R5 is hydrogen, C1-C6alkyl, C1-C6heteroalkyl, -F, -Cl, -Br, or phenyl, wherein each of the alkyl, heteroalkyl, and phenyl is optionally substituted with one or more substituents selected from C1-C3alkyl, C1-C3alkoxy, -F, -Cl, -Br, and -CN. In some embodiments, R5 is hydrogen. In some embodiments, R5 is C1-C6alkyl, C1-C6heteroalkyl, -F, -Cl, -Br, or phenyl, wherein each of the alkyl, heteroalkyl, and phenyl is optionally substituted with one or more substituents selected from C1-C3alkyl, C1- C3alkoxy, -F, -Cl, -Br, and -CN. In some embodiments, R5is C1-C6alkyl, C1-C6heteroalkyl, -F, -Cl, -Br, or phenyl, wherein each of the alkyl, heteroalkyl, and phenyl is optionally substituted with one or more substituents selected from methyl, -O-CH3, -F, -Cl, -Br, and -CN. [0054] In some embodiments of Formula (I), (I-A-8), (I-A-9), or (I-A-10),R8 is hydrogen or C1-C6alkyl. In some embodiments, R8 is hydrogen or C1-C3alkyl. In some embodiments, R8 is hydrogen. In some embodiments, R8 is C1-C3alkyl. [0055] In some embodiments of Formula (I), (I-A-8), (I-A-9), or (I-A-10), R9 is hydrogen or C1-C3alkyl. In some embodiments, R9 is hydrogen. In some embodiments, R9 is -CH3. In some embodiments, R9 is - CH2CH3. [0056] In some embodiments of Formula (I), (I-A-8), (I-A-9), or (I-A-10), R8 and R9 are taken together to form a heterocycloalkyl, wherein the heterocycloalkyl is optionally substituted with one or more substituents selected from C1-C3alkyl, C1-C3haloalkyl, C1-C3alkoxy, halogen, -OH, and =O. In some embodiments, the heterocycloalkyl is a 5- to 6- membered heterocycloalkyl optionally substituted with one or more substituents selected from C1-C3alkyl, C1-C3haloalkyl, C1-C3alkoxy, halogen, -OH, and =O. In some embodiments, R8 and R9 are taken together to form a pyrrolidinyl, an imidazolidinyl, a piperidinyl, a piperazinyl, or a morpholino, each of which is optionally substituted with one or more substituents selected from C1-C3alkyl, C1-C3haloalkyl, C1-C3alkoxy, halogen, -OH, and =O. In some embodiments, R8 and R9 are taken together to form a pyrrolidinyl. In some embodiments, R8 and R9 are taken together to form an imidazolidinyl. In some embodiments, R8 and R9 are taken together to form a piperidinyl. In some embodiments, R8 and R9 are taken together to form a piperazinyl. In some embodiments, R8 and R8 are taken together to form a morpholino. [0057] In some embodiments, a compound disclosed herein, or a pharmaceutically acceptable salt, pharmaceutically acceptable solvate, diastereomeric mixture, individual enantiomer, or isotopically enriched
[0051] In some embodiments of Formula (I), (I-A-2), (I-A-5), (I-A-8), (I-A-9), (I-A-11), (I-A-14), (I-A-15), (I-A-17), (I-A-18), or (I-A-20), R3 is hydrogen, C1-C6alkyl, C1-C6heteroalkyl, -F, -Cl, -Br, or phenyl, wherein each of the alkyl, heteroalkyl, and phenyl is optionally substituted with one or more substituents selected from C1-C3alkyl, C1-C3alkoxy, -F, -Cl, -Br, and -CN. In some embodiments, R3 is hydrogen. In some embodiments, R3 is C1-C6alkyl, C1-C6heteroalkyl, -F, -Cl, -Br, or phenyl, wherein each of the alkyl, heteroalkyl, and phenyl is optionally substituted with one or more substituents selected from C1-C3alkyl, C1- C3alkoxy, -F, -Cl, -Br, and -CN. In some embodiments, R3 is C1-C6alkyl, C1-C6heteroalkyl, -F, -Cl, -Br, or phenyl, wherein each of the alkyl, heteroalkyl, and phenyl is optionally substituted with one or more substituents selected from methyl, -O-CH3, -F, -Cl, -Br, and -CN. [0052] In some embodiments of Formula (I), (I-A-3), (I-A-6), (I-A-8), (I-A-10), (I-A-12), (I-A-14), (I-A- 16), (I-A-17), (I-A-19), or (I-A-20), R4 is hydrogen, C1-C6alkyl, C1-C6heteroalkyl, -F, -Cl, -Br, or phenyl, wherein each of the alkyl, heteroalkyl, and phenyl is optionally substituted with one or more substituents selected from C1-C3alkyl, C1-C3alkoxy, -F, -Cl, -Br, and -CN. In some embodiments, R4 is hydrogen. In some embodiments, R4 is C1-C6alkyl, C1-C6heteroalkyl, -F, -Cl, -Br, or phenyl, wherein each of the alkyl, heteroalkyl, and phenyl is optionally substituted with one or more substituents selected from C1-C3alkyl, C1- C3alkoxy, -F, -Cl, -Br, and -CN. In some embodiments, R4 is C1-C6alkyl, C1-C6heteroalkyl, -F, -Cl, -Br, or phenyl, wherein each of the alkyl, heteroalkyl, and phenyl is optionally substituted with one or more substituents selected from methyl, -O-CH3, -F, -Cl, -Br, and -CN.
WSGR Docket No.55776-726.601 [0053] In some embodiments of Formula (I), (I-A-4), (I-A-7), (I-A-9), (I-A-10), (I-A-13), (I-A-15), (I-A- 16), (I-A-18), (I-A-19), or (I-A-20), R5 is hydrogen, C1-C6alkyl, C1-C6heteroalkyl, -F, -Cl, -Br, or phenyl, wherein each of the alkyl, heteroalkyl, and phenyl is optionally substituted with one or more substituents selected from C1-C3alkyl, C1-C3alkoxy, -F, -Cl, -Br, and -CN. In some embodiments, R5 is hydrogen. In some embodiments, R5 is C1-C6alkyl, C1-C6heteroalkyl, -F, -Cl, -Br, or phenyl, wherein each of the alkyl, heteroalkyl, and phenyl is optionally substituted with one or more substituents selected from C1-C3alkyl, C1- C3alkoxy, -F, -Cl, -Br, and -CN. In some embodiments, R5is C1-C6alkyl, C1-C6heteroalkyl, -F, -Cl, -Br, or phenyl, wherein each of the alkyl, heteroalkyl, and phenyl is optionally substituted with one or more substituents selected from methyl, -O-CH3, -F, -Cl, -Br, and -CN. [0054] In some embodiments of Formula (I), (I-A-8), (I-A-9), or (I-A-10),R8 is hydrogen or C1-C6alkyl. In some embodiments, R8 is hydrogen or C1-C3alkyl. In some embodiments, R8 is hydrogen. In some embodiments, R8 is C1-C3alkyl. [0055] In some embodiments of Formula (I), (I-A-8), (I-A-9), or (I-A-10), R9 is hydrogen or C1-C3alkyl. In some embodiments, R9 is hydrogen. In some embodiments, R9 is -CH3. In some embodiments, R9 is - CH2CH3. [0056] In some embodiments of Formula (I), (I-A-8), (I-A-9), or (I-A-10), R8 and R9 are taken together to form a heterocycloalkyl, wherein the heterocycloalkyl is optionally substituted with one or more substituents selected from C1-C3alkyl, C1-C3haloalkyl, C1-C3alkoxy, halogen, -OH, and =O. In some embodiments, the heterocycloalkyl is a 5- to 6- membered heterocycloalkyl optionally substituted with one or more substituents selected from C1-C3alkyl, C1-C3haloalkyl, C1-C3alkoxy, halogen, -OH, and =O. In some embodiments, R8 and R9 are taken together to form a pyrrolidinyl, an imidazolidinyl, a piperidinyl, a piperazinyl, or a morpholino, each of which is optionally substituted with one or more substituents selected from C1-C3alkyl, C1-C3haloalkyl, C1-C3alkoxy, halogen, -OH, and =O. In some embodiments, R8 and R9 are taken together to form a pyrrolidinyl. In some embodiments, R8 and R9 are taken together to form an imidazolidinyl. In some embodiments, R8 and R9 are taken together to form a piperidinyl. In some embodiments, R8 and R9 are taken together to form a piperazinyl. In some embodiments, R8 and R8 are taken together to form a morpholino. [0057] In some embodiments, a compound disclosed herein, or a pharmaceutically acceptable salt, pharmaceutically acceptable solvate, diastereomeric mixture, individual enantiomer, or isotopically enriched
 , , or . [0058] In some embodiments, a compound disclosed herein, or a pharmaceutically acceptable salt, pharmaceutically acceptable solvate, diastereomeric mixture, individual enantiomer, or isotopically enriched
WSGR Docket No.55776-726.601 , ,
, , or . [0058] In some embodiments, a compound disclosed herein, or a pharmaceutically acceptable salt, pharmaceutically acceptable solvate, diastereomeric mixture, individual enantiomer, or isotopically enriched
WSGR Docket No.55776-726.601 , ,
 [0109] Step 1: Synthesis of RP-1 (7-bromo-4,5-difluoro-1H-indole):
WSGR Docket No.55776-726.601 [0110] To a stirred
[0109] Step 1: Synthesis of RP-1 (7-bromo-4,5-difluoro-1H-indole):
WSGR Docket No.55776-726.601 [0110] To a stirred
 mmol, 1.0 eq) in THF (190 mL) at -78 °C was added a vinyl magnesium bromide solution (1M in THF, 32.2 g, 319 mmol, 4.0 eq). The reaction mixture was allowed to warm to room temperature and was stirred at that temperature for 12 h. The reaction mixture was quenched with an aqueous NH4Cl solution and extracted with multiple portions of EtOAc. The combined organic extracts were dried over anhydrous Na2SO4, the solids were removed by filtration, and the filtrate was concentrated in vacuo to yield crude product. The crude material was purified by silica gel chromatography to afford 7-bromo-4,5-difluoro-1H-indole (RP-1) (6.2 g, 33%) as a brown liquid. ESI-MS m/z: 231.8 [M-H]+. [0111] Step 2: Synthesis of RP-2 (1-allyl-7-bromo-4,5-difluoro-1H-indole):
mmol, 1.0 eq) in THF (190 mL) at -78 °C was added a vinyl magnesium bromide solution (1M in THF, 32.2 g, 319 mmol, 4.0 eq). The reaction mixture was allowed to warm to room temperature and was stirred at that temperature for 12 h. The reaction mixture was quenched with an aqueous NH4Cl solution and extracted with multiple portions of EtOAc. The combined organic extracts were dried over anhydrous Na2SO4, the solids were removed by filtration, and the filtrate was concentrated in vacuo to yield crude product. The crude material was purified by silica gel chromatography to afford 7-bromo-4,5-difluoro-1H-indole (RP-1) (6.2 g, 33%) as a brown liquid. ESI-MS m/z: 231.8 [M-H]+. [0111] Step 2: Synthesis of RP-2 (1-allyl-7-bromo-4,5-difluoro-1H-indole):
 [0112] To a stirred solution of RP-1 (6.2 g, 26.7 mmol, 1.0 eq) in DMF (62 mL) at 0 ℃ was added NaH (60% in mineral oil, 1.28 g, 53.4 mmol, 2.0 eq). The reaction mixture was stirred for 20 min at that temperature and allyl bromide (3.87 g, 32 mmol, 1.2 eq) was added. The reaction mixture was warmed to room temperature and stirred for 3 h. The reaction was diluted with ice cold water and extracted with multiple portions of EtOAc. The combined organic extracts were dried over anhydrous Na2SO4, the solids were removed by filtration, and the filtrate was concentrated in vacuo to yield crude product. The crude material was purified by silica gel chromatography to afford 1-allyl-7-bromo-4,5-difluoro-1H-indole (RP-2) (6.5 g, 90%) as a brown liquid. ESI-MS m/z: 290.1 [M+H2O]+. [0113] Step 3: RP-
[0112] To a stirred solution of RP-1 (6.2 g, 26.7 mmol, 1.0 eq) in DMF (62 mL) at 0 ℃ was added NaH (60% in mineral oil, 1.28 g, 53.4 mmol, 2.0 eq). The reaction mixture was stirred for 20 min at that temperature and allyl bromide (3.87 g, 32 mmol, 1.2 eq) was added. The reaction mixture was warmed to room temperature and stirred for 3 h. The reaction was diluted with ice cold water and extracted with multiple portions of EtOAc. The combined organic extracts were dried over anhydrous Na2SO4, the solids were removed by filtration, and the filtrate was concentrated in vacuo to yield crude product. The crude material was purified by silica gel chromatography to afford 1-allyl-7-bromo-4,5-difluoro-1H-indole (RP-2) (6.5 g, 90%) as a brown liquid. ESI-MS m/z: 290.1 [M+H2O]+. [0113] Step 3: RP-
 [0114] To a stirred solution of RP-2 (2.0 g, 7.35 mmol, 1.0 eq) in DMF (20 mL) was added K2CO3 (2.0 g, 14.7 mmol, 2.0 eq) at room temperature and the reaction was degassed under N2 atmosphere for 20 minutes. To this reaction mixture was added TBAI (4.06 g, 11 mmol, 1.5 eq) followed by Pd(OAc)2 (0.33 g, 1.47 mmol, 0.2 eq). The reaction mixture was heated at 80 ℃ and was stirred at that temperature for 3 h. The reaction mixture was cooled to room temperature, diluted with water (60 mL), filtered through a pad of
WSGR Docket No.55776-726.601 celite, and the filter pad was washed with EtOAc. The combined organic phase was dried over anhydrous Na2SO4, the solids were removed by filtration, and the filtrate was concentrated in vacuo to obtain the crude. The crude was purified by silica gel chromatography column to afford RP-3 (0.75 g, 53%) as a mixture of regio-isomers. ESI-MS m/z:190.2 [M+H]+. [0115] Step 4: Synthesis of RP-4 (8,9-difluoro-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5-ol):
[0114] To a stirred solution of RP-2 (2.0 g, 7.35 mmol, 1.0 eq) in DMF (20 mL) was added K2CO3 (2.0 g, 14.7 mmol, 2.0 eq) at room temperature and the reaction was degassed under N2 atmosphere for 20 minutes. To this reaction mixture was added TBAI (4.06 g, 11 mmol, 1.5 eq) followed by Pd(OAc)2 (0.33 g, 1.47 mmol, 0.2 eq). The reaction mixture was heated at 80 ℃ and was stirred at that temperature for 3 h. The reaction mixture was cooled to room temperature, diluted with water (60 mL), filtered through a pad of
WSGR Docket No.55776-726.601 celite, and the filter pad was washed with EtOAc. The combined organic phase was dried over anhydrous Na2SO4, the solids were removed by filtration, and the filtrate was concentrated in vacuo to obtain the crude. The crude was purified by silica gel chromatography column to afford RP-3 (0.75 g, 53%) as a mixture of regio-isomers. ESI-MS m/z:190.2 [M+H]+. [0115] Step 4: Synthesis of RP-4 (8,9-difluoro-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5-ol):
 [0118] To a stirred solution of RP-4 (170 mg, 0.81 mmol, 1.0 eq) in dichloromethane (10 mL) was added triethylamine (163 mg, 1.62 mmol, 2.0 eq) at 0 ℃ and the reaction mixture was stirred for 10 mins. To the reaction mixture was added TsCl (229 mg, 1.21 mmol, 1.5 eq), followed by DMAP (9.92 mg, 0.08 mmol, 0.1 eq). The reaction mixture was warmed to room temperature and stirred at that temperature for 6 h. The reaction mixture was diluted with ice cold water and extracted with multiple portions of EtOAc. The combined organic layers were washed with an aqueous solution of NaHCO3, dried over anhydrous Na2SO4, the solids were removed by filtration, and the filtrate was concentrated in vacuo to obtain crude product. The crude material was purified by silica gel chromatography to afford RP-5 (8,9-difluoro-5,6-dihydro-4H- pyrrolo[3,2,1-ij]quinolin-5-yl 4-methylbenzenesulfonate) (110 mg, 37%) as a brown liquid, which was carried forward without further purification. 1H NMR (400 MHz, CHLOROFORM-d) δ: 7.73 (d, J = 7.8
WSGR Docket No.55776-726.601 Hz, 2H), 7.34 (d, J = 7.8 Hz, 2H), 6.98 (d, J = 2.4 Hz, 1H), 6.66 (dd, J = 6.8, 10 Hz, 1H), 6.54 (br s, 1H), 5.35 - 5.11 (m, 1H), 4.29 (d, J = 3.4 Hz, 2H), 3.10 (s, 2H), 2.47 (s, 3H) ppm. [0119] Step 6: Synthesis of RP-6 (5-azido-8,9-difluoro-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinoline):
[0118] To a stirred solution of RP-4 (170 mg, 0.81 mmol, 1.0 eq) in dichloromethane (10 mL) was added triethylamine (163 mg, 1.62 mmol, 2.0 eq) at 0 ℃ and the reaction mixture was stirred for 10 mins. To the reaction mixture was added TsCl (229 mg, 1.21 mmol, 1.5 eq), followed by DMAP (9.92 mg, 0.08 mmol, 0.1 eq). The reaction mixture was warmed to room temperature and stirred at that temperature for 6 h. The reaction mixture was diluted with ice cold water and extracted with multiple portions of EtOAc. The combined organic layers were washed with an aqueous solution of NaHCO3, dried over anhydrous Na2SO4, the solids were removed by filtration, and the filtrate was concentrated in vacuo to obtain crude product. The crude material was purified by silica gel chromatography to afford RP-5 (8,9-difluoro-5,6-dihydro-4H- pyrrolo[3,2,1-ij]quinolin-5-yl 4-methylbenzenesulfonate) (110 mg, 37%) as a brown liquid, which was carried forward without further purification. 1H NMR (400 MHz, CHLOROFORM-d) δ: 7.73 (d, J = 7.8
WSGR Docket No.55776-726.601 Hz, 2H), 7.34 (d, J = 7.8 Hz, 2H), 6.98 (d, J = 2.4 Hz, 1H), 6.66 (dd, J = 6.8, 10 Hz, 1H), 6.54 (br s, 1H), 5.35 - 5.11 (m, 1H), 4.29 (d, J = 3.4 Hz, 2H), 3.10 (s, 2H), 2.47 (s, 3H) ppm. [0119] Step 6: Synthesis of RP-6 (5-azido-8,9-difluoro-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinoline):
 [0120] To a stirred solution mg, mL) at 0 ℃ was added NaN3 (28.1 mg, 0.43 mmol, 1.5 eq). The reaction mixture was allowed to warm to room temperature, was heated to 65 ℃, and was stirred at that temperature for 2 h. The reaction mixture was diluted with ice cold water and extracted with multiple portions of diethyl ether. The combined organic layers were washed with ice cold water, followed by an aqueous solution of NaCl. The organic phase was dried over anhydrous Na2SO4, the solids were filtered, and the filtrate was concentrated in vacuo to afford crude 5-azido-8,9-difluoro-5,6- dihydro-4H-pyrrolo[3,2,1-ij]quinoline (RP-6) (60 mg, 83%). The crude was directly used in the next step without purification. ESI-MS m/z: 235.3 [M+H]+. [0121] Step 7: Synthesis of Compound 9 (8,9-difluoro-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5- amine)
[0120] To a stirred solution mg, mL) at 0 ℃ was added NaN3 (28.1 mg, 0.43 mmol, 1.5 eq). The reaction mixture was allowed to warm to room temperature, was heated to 65 ℃, and was stirred at that temperature for 2 h. The reaction mixture was diluted with ice cold water and extracted with multiple portions of diethyl ether. The combined organic layers were washed with ice cold water, followed by an aqueous solution of NaCl. The organic phase was dried over anhydrous Na2SO4, the solids were filtered, and the filtrate was concentrated in vacuo to afford crude 5-azido-8,9-difluoro-5,6- dihydro-4H-pyrrolo[3,2,1-ij]quinoline (RP-6) (60 mg, 83%). The crude was directly used in the next step without purification. ESI-MS m/z: 235.3 [M+H]+. [0121] Step 7: Synthesis of Compound 9 (8,9-difluoro-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5- amine)
 [0122] To a stirred solution of crude RP-6 (60 mg, 0.24 mmol, 1.0 eq) in THF (0.6 mL) and H2O (0.3 mL) at 0 ℃ was added TPP (94.6 mg, 0.36 mmol, 1.5 eq). The reaction mixture was allowed to warm to room temperature, was heated to 80 ℃, and was stirred at that temperature for 2 h. The reaction mixture was allowed to cool at room temperature, diluted with ice cold water, and extracted with multiple portions of EtOAc. The combined organic extracts were washed with an aqueous solution of NaCl, dried over anhydrous Na2SO4, the solids were filtered, and the filtrate was concentrated in vacuo to obtain crude material. The crude product was purified by silica gel chromatography to afford 8,9-difluoro-5,6-dihydro- 4H-pyrrolo[3,2,1-ij]quinolin-5-amine (Compound 9) (30 mg, 56 %). ESI-MS m/z: 209.0 [M+H]+. [0123] Step 8: Synthesis of Compound 12 (8,9-difluoro-N,N-dimethyl-5,6-dihydro-4H-pyrrolo[3,2,1- ij]quinolin-5-amine):
WSGR Docket No.55776-726.601
[0122] To a stirred solution of crude RP-6 (60 mg, 0.24 mmol, 1.0 eq) in THF (0.6 mL) and H2O (0.3 mL) at 0 ℃ was added TPP (94.6 mg, 0.36 mmol, 1.5 eq). The reaction mixture was allowed to warm to room temperature, was heated to 80 ℃, and was stirred at that temperature for 2 h. The reaction mixture was allowed to cool at room temperature, diluted with ice cold water, and extracted with multiple portions of EtOAc. The combined organic extracts were washed with an aqueous solution of NaCl, dried over anhydrous Na2SO4, the solids were filtered, and the filtrate was concentrated in vacuo to obtain crude material. The crude product was purified by silica gel chromatography to afford 8,9-difluoro-5,6-dihydro- 4H-pyrrolo[3,2,1-ij]quinolin-5-amine (Compound 9) (30 mg, 56 %). ESI-MS m/z: 209.0 [M+H]+. [0123] Step 8: Synthesis of Compound 12 (8,9-difluoro-N,N-dimethyl-5,6-dihydro-4H-pyrrolo[3,2,1- ij]quinolin-5-amine):
WSGR Docket No.55776-726.601
 [0126] To a stirred solution of Compound 9 (1.1 g, 5.28 mmol, 1.0 eq) in DCM (15 mL) at 0 ℃ was added triethylamine (1.46 mL, 10.5 mmol, 2.0 eq), followed by Boc2O (969 µL, 4.22 mmol, 0.8 eq), and DMAP (cat). The resulting reaction mixture was slowly warmed to room temperature and stirred at that temperature for 16 h. The reaction mixture was diluted with water and extracted with multiple portions of DCM. The combined organic phase was washed with brine, dried over Na2SO4, the solids were removed by filtration, and the filtrate was concentrated in vacuo to afford crude material. The crude compound was purified by silica gel chromatography to afford RP-8 (1.1 g, 68%) as an off white solid.1H NMR (400 MHz, CHLOROFORM-d) δ: 7.08 (s, 1H), 6.81 (dd, J = 6.8, 10.4 Hz, 1H), 6.56 (d, J = 2.6 Hz, 1H), 4.52 (s, 2H), 4.28 - 4.21 (m, 1H), 4.14 (d, J = 9.6 Hz, 1H), 3.19 (d, J = 16.2 Hz, 1H), 2.92 (d, J = 15.4 Hz, 1H), 1.42 (br s, 9H) ppm. ESI-MS m/z: 309.2 [M+H]+. [0127] 1.1 g of RP-8 was subjected to chiral separation to get 460 mg of RP-9 (tert-butyl (R)-(8,9-difluoro- 5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5-yl)carbamate)& 450 mg of RP-10 (tert-butyl (S)-(8,9-difluoro- 5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5-yl)carbamate).
WSGR Docket No.55776-726.601 Column : Chiralpak-AD-H(250mm, 30mm, 5µ) Mobile Phase A : 0.1% DEA in n- Hexane Mobile Phase B : EtOH Flow rate : 36.0 mL/min [0128] RP-9: 1H NMR (400 MHz, CHLOROFORM-d) δ = 7.08 (d, J = 2.4 Hz, 1H), 6.81 (dd, J = 6.6, 10.4 Hz, 1H), 6.56 (d, J = 2.9 Hz, 1H), 4.59 - 4.48 (m, 1H), 4.27 - 4.21 (m, 1H), 4.18 - 4.10 (m, 1H), 3.19 (d, J = 16.2 Hz, 1H), 2.96 - 2.89 (m, 1H), 1.41 (br s, 9H) ppm. ESI-MS m/z: 308.7 [M+H]+. [0129] RP-10: 1H NMR (400 MHz, CHLOROFORM-d) δ = 7.08 (d, J = 2.4 Hz, 1H), 6.81 (dd, J = 6.4, 10.8 Hz, 1H), 6.56 (d, J = 2.9 Hz, 1H), 4.53 (d, J = 10.2 Hz, 2H), 4.28 - 4.20 (m, 1H), 4.14 (2 d, J = 9.8 Hz, 1H), 3.19 (d, J = 16.2 Hz, 1H), 2.97 - 2.88 (m, 1H), 1.41 (br s, 9H) ppm. ESI-MS m/z: 308.9 [M+H]+. [0130] Step-10: Synthesis of Compound 10 ((R)-8,9-difluoro-5,6-dihydro-4H-pyrrolo[3,2,1- ij]quinolin-5-amine) & Compound 11 ((S)-8,9-difluoro-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5- amine):
[0126] To a stirred solution of Compound 9 (1.1 g, 5.28 mmol, 1.0 eq) in DCM (15 mL) at 0 ℃ was added triethylamine (1.46 mL, 10.5 mmol, 2.0 eq), followed by Boc2O (969 µL, 4.22 mmol, 0.8 eq), and DMAP (cat). The resulting reaction mixture was slowly warmed to room temperature and stirred at that temperature for 16 h. The reaction mixture was diluted with water and extracted with multiple portions of DCM. The combined organic phase was washed with brine, dried over Na2SO4, the solids were removed by filtration, and the filtrate was concentrated in vacuo to afford crude material. The crude compound was purified by silica gel chromatography to afford RP-8 (1.1 g, 68%) as an off white solid.1H NMR (400 MHz, CHLOROFORM-d) δ: 7.08 (s, 1H), 6.81 (dd, J = 6.8, 10.4 Hz, 1H), 6.56 (d, J = 2.6 Hz, 1H), 4.52 (s, 2H), 4.28 - 4.21 (m, 1H), 4.14 (d, J = 9.6 Hz, 1H), 3.19 (d, J = 16.2 Hz, 1H), 2.92 (d, J = 15.4 Hz, 1H), 1.42 (br s, 9H) ppm. ESI-MS m/z: 309.2 [M+H]+. [0127] 1.1 g of RP-8 was subjected to chiral separation to get 460 mg of RP-9 (tert-butyl (R)-(8,9-difluoro- 5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5-yl)carbamate)& 450 mg of RP-10 (tert-butyl (S)-(8,9-difluoro- 5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5-yl)carbamate).
WSGR Docket No.55776-726.601 Column : Chiralpak-AD-H(250mm, 30mm, 5µ) Mobile Phase A : 0.1% DEA in n- Hexane Mobile Phase B : EtOH Flow rate : 36.0 mL/min [0128] RP-9: 1H NMR (400 MHz, CHLOROFORM-d) δ = 7.08 (d, J = 2.4 Hz, 1H), 6.81 (dd, J = 6.6, 10.4 Hz, 1H), 6.56 (d, J = 2.9 Hz, 1H), 4.59 - 4.48 (m, 1H), 4.27 - 4.21 (m, 1H), 4.18 - 4.10 (m, 1H), 3.19 (d, J = 16.2 Hz, 1H), 2.96 - 2.89 (m, 1H), 1.41 (br s, 9H) ppm. ESI-MS m/z: 308.7 [M+H]+. [0129] RP-10: 1H NMR (400 MHz, CHLOROFORM-d) δ = 7.08 (d, J = 2.4 Hz, 1H), 6.81 (dd, J = 6.4, 10.8 Hz, 1H), 6.56 (d, J = 2.9 Hz, 1H), 4.53 (d, J = 10.2 Hz, 2H), 4.28 - 4.20 (m, 1H), 4.14 (2 d, J = 9.8 Hz, 1H), 3.19 (d, J = 16.2 Hz, 1H), 2.97 - 2.88 (m, 1H), 1.41 (br s, 9H) ppm. ESI-MS m/z: 308.9 [M+H]+. [0130] Step-10: Synthesis of Compound 10 ((R)-8,9-difluoro-5,6-dihydro-4H-pyrrolo[3,2,1- ij]quinolin-5-amine) & Compound 11 ((S)-8,9-difluoro-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5- amine):
 WSGR Docket No.55776-726.601 [0132] To a stirred solution of RP-10 (450 mg, 1.45 mol, 1.0 eq) in DCM (15 mL) at 0 ℃ were added 2,6- lutidine (837 µL, 7.24 mmol, 5.0 eq) and TMSOTf (1.04 mL, 5.80 mmol, 4.0 eq). The reaction mixture slowly warmed to room temperature and was stirred at that temperature for 4 h. The reaction mixture was quenched with a saturated aqueous NaHCO3 solution and extracted with multiple portions of DCM. The combined organic phase was dried over Na2SO4, the solids were filtered, and the filtrate was concentrated in vacuo to yield crude product. The crude material was purified by silica gel chromatography to afford Compound 11 (240 mg, 80%). 1H NMR (400 MHz, DMSO-d6) δ: 7.42 (d, J = 2.9 Hz, 1H), 6.91 (dd, J = 6.8, 11.2 Hz, 1H), 6.46 (d, J = 2.4 Hz, 1H), 4.24 (dd, J = 3.6, 12.0 Hz, 1H), 4.04 (s, 2H), 3.81 (dd, J = 7.8, 12.2 Hz, 1H), 3.52 - 3.47 (m, 1H), 3.05 (dd, J = 3.4, 16.0 Hz, 1H), 2.76 - 2.70 (m, 1H) ppm. ESI-MS m/z: 209.3 [M+H]+. [0133] Step-11; Synthesis of Compound 13 ((R)-8,9-difluoro-N,N-dimethyl-5,6-dihydro-4H- pyrrolo[3,2,1-ij]quinolin-5-amine) & Compound 14 ((S)-8,9-difluoro-N,N-dimethyl-5,6-dihydro-4H- pyrrolo[3,2,1-ij]quinolin-5-amine)
WSGR Docket No.55776-726.601 [0132] To a stirred solution of RP-10 (450 mg, 1.45 mol, 1.0 eq) in DCM (15 mL) at 0 ℃ were added 2,6- lutidine (837 µL, 7.24 mmol, 5.0 eq) and TMSOTf (1.04 mL, 5.80 mmol, 4.0 eq). The reaction mixture slowly warmed to room temperature and was stirred at that temperature for 4 h. The reaction mixture was quenched with a saturated aqueous NaHCO3 solution and extracted with multiple portions of DCM. The combined organic phase was dried over Na2SO4, the solids were filtered, and the filtrate was concentrated in vacuo to yield crude product. The crude material was purified by silica gel chromatography to afford Compound 11 (240 mg, 80%). 1H NMR (400 MHz, DMSO-d6) δ: 7.42 (d, J = 2.9 Hz, 1H), 6.91 (dd, J = 6.8, 11.2 Hz, 1H), 6.46 (d, J = 2.4 Hz, 1H), 4.24 (dd, J = 3.6, 12.0 Hz, 1H), 4.04 (s, 2H), 3.81 (dd, J = 7.8, 12.2 Hz, 1H), 3.52 - 3.47 (m, 1H), 3.05 (dd, J = 3.4, 16.0 Hz, 1H), 2.76 - 2.70 (m, 1H) ppm. ESI-MS m/z: 209.3 [M+H]+. [0133] Step-11; Synthesis of Compound 13 ((R)-8,9-difluoro-N,N-dimethyl-5,6-dihydro-4H- pyrrolo[3,2,1-ij]quinolin-5-amine) & Compound 14 ((S)-8,9-difluoro-N,N-dimethyl-5,6-dihydro-4H- pyrrolo[3,2,1-ij]quinolin-5-amine)
 [0134] To a stirred solution of Compound 10 (200 mg, 960 µmol, 1.0 eq) in THF/MeOH mixture (1:1, 10 vol) was added a solution of formaldehyde (37% in water, 233 mg, 2.88 mmol, 3.0 eq). The reaction mixture was stirred for 30 minutes, then was cooled to 0 ℃, and NaCNBH3 (120 mg, 1.91 mmol, 2.0 eq) was added. The reaction mixture was warmed to room temperature and was stirred at that temperature for 12 h. The volatiles were evaporated in vacuo, the obtained crude residue was diluted with water (10 mL), and the entire mixture was extracted twice with a 10% MeOH/DCM solution. The combined organic phase was dried over Na2SO4, the solids were filtered, and the filtrate was concentrated in vacuo to afford crude compound. The crude material was purified by silica gel chromatography to afford Compound 13 (120 mg, 53%).
[0134] To a stirred solution of Compound 10 (200 mg, 960 µmol, 1.0 eq) in THF/MeOH mixture (1:1, 10 vol) was added a solution of formaldehyde (37% in water, 233 mg, 2.88 mmol, 3.0 eq). The reaction mixture was stirred for 30 minutes, then was cooled to 0 ℃, and NaCNBH3 (120 mg, 1.91 mmol, 2.0 eq) was added. The reaction mixture was warmed to room temperature and was stirred at that temperature for 12 h. The volatiles were evaporated in vacuo, the obtained crude residue was diluted with water (10 mL), and the entire mixture was extracted twice with a 10% MeOH/DCM solution. The combined organic phase was dried over Na2SO4, the solids were filtered, and the filtrate was concentrated in vacuo to afford crude compound. The crude material was purified by silica gel chromatography to afford Compound 13 (120 mg, 53%).
 [0135] To a stirred solution of Compound 11 (200 mg, 960 µmol, 1.0 eq) in THF/MeOH mixture (1:1, 10 vol) was added a solution of formaldehyde (37% in water, 233 mg, 2.88 mmol, 3.0 eq). The reaction
WSGR Docket No.55776-726.601 mixture was stirred for 30 minutes, then was cooled to 0 ℃, and NaCNBH3 (120 mg, 1.91 mmol, 2.0 eq) was added. The reaction mixture was warmed to room temperature and was stirred at that temperature for 12 h. The volatiles were evaporated in vacuo, the obtained crude residue was diluted with water (10 mL), and the entire mixture was extracted twice with a 10% MeOH/DCM solution. The combined organic phase was dried over Na2SO4, the solids were filtered, and the filtrate was concentrated in vacuo to afford crude compound. The crude material was purified by silica gel chromatography to afford Compound 14 (105 mg, 46%). Example A2: [0136] Scheme-2: Synthesis of Intermediates GG-6a, GG-6b, GG-6c, and GG-6d
[0135] To a stirred solution of Compound 11 (200 mg, 960 µmol, 1.0 eq) in THF/MeOH mixture (1:1, 10 vol) was added a solution of formaldehyde (37% in water, 233 mg, 2.88 mmol, 3.0 eq). The reaction
WSGR Docket No.55776-726.601 mixture was stirred for 30 minutes, then was cooled to 0 ℃, and NaCNBH3 (120 mg, 1.91 mmol, 2.0 eq) was added. The reaction mixture was warmed to room temperature and was stirred at that temperature for 12 h. The volatiles were evaporated in vacuo, the obtained crude residue was diluted with water (10 mL), and the entire mixture was extracted twice with a 10% MeOH/DCM solution. The combined organic phase was dried over Na2SO4, the solids were filtered, and the filtrate was concentrated in vacuo to afford crude compound. The crude material was purified by silica gel chromatography to afford Compound 14 (105 mg, 46%). Example A2: [0136] Scheme-2: Synthesis of Intermediates GG-6a, GG-6b, GG-6c, and GG-6d
 [0137] Synthesis of Intermediate Compound GG-6a: tert-butyl (5,6-dihydro-4H-pyrrolo[3,2,1- ij]quinolin-5-yl)carbamate
[0137] Synthesis of Intermediate Compound GG-6a: tert-butyl (5,6-dihydro-4H-pyrrolo[3,2,1- ij]quinolin-5-yl)carbamate
 [0138] Step-1a: Synthesis of GG-1a: methyl 2-((tert-butoxycarbonyl)amino)-3-(1H-indol-7-yl) propanoate):
WSGR Docket No.55776-726.601
[0138] Step-1a: Synthesis of GG-1a: methyl 2-((tert-butoxycarbonyl)amino)-3-(1H-indol-7-yl) propanoate):
WSGR Docket No.55776-726.601
 [0139] To a stirred solution of 7-bromo-1H-indole (4.0 g, 20.4 mmol, 1.0 eq) in DMF (100 mL) at room temperature were added Compound GG-2, methyl 2-((tert-butoxycarbonyl)amino)-3-iodopropanoate (6.71 g, 20.4 mmol 1.0 eq) and Zn dust (5.33 g, 81.6 mmol, 4.0 eq). The resulting mixture was purged with a nitrogen atmosphere and then Pd(amphos)2Cl2 (715 mg, 1.01 mmol, 0.05 eq) was added. The reaction mixture was stirred at ambient temperature for 12 hours. The reaction was diluted with ice-water (50 mL) and extracted with EtOAc (2 x 30 mL). The combined organic layers were washed with an aqueous solution of NaCl (10 mL), the organic layer was dried over anhydrous Na2SO4, solids were removed by filtration, and the filtrate was concentrated in vacuo. The crude material was purified by silica gel chromatography (12% EtOAc/Hexane), to afford methyl 2-((tert-butoxycarbonyl)amino)-3-(1H-indol-7-yl) propanoate), Compound GG-3a (3.8 g, 59% yield) as a pale yellow solid. ESI-MS m/z: 318.9 [M-H]-. [0140] Step-2a: Synthesis of Compound GG-4a: tert-butyl (1-hydroxy-3-(1H-indol-7-yl)propan-2- yl)carbamate
[0139] To a stirred solution of 7-bromo-1H-indole (4.0 g, 20.4 mmol, 1.0 eq) in DMF (100 mL) at room temperature were added Compound GG-2, methyl 2-((tert-butoxycarbonyl)amino)-3-iodopropanoate (6.71 g, 20.4 mmol 1.0 eq) and Zn dust (5.33 g, 81.6 mmol, 4.0 eq). The resulting mixture was purged with a nitrogen atmosphere and then Pd(amphos)2Cl2 (715 mg, 1.01 mmol, 0.05 eq) was added. The reaction mixture was stirred at ambient temperature for 12 hours. The reaction was diluted with ice-water (50 mL) and extracted with EtOAc (2 x 30 mL). The combined organic layers were washed with an aqueous solution of NaCl (10 mL), the organic layer was dried over anhydrous Na2SO4, solids were removed by filtration, and the filtrate was concentrated in vacuo. The crude material was purified by silica gel chromatography (12% EtOAc/Hexane), to afford methyl 2-((tert-butoxycarbonyl)amino)-3-(1H-indol-7-yl) propanoate), Compound GG-3a (3.8 g, 59% yield) as a pale yellow solid. ESI-MS m/z: 318.9 [M-H]-. [0140] Step-2a: Synthesis of Compound GG-4a: tert-butyl (1-hydroxy-3-(1H-indol-7-yl)propan-2- yl)carbamate
 [0143] To a stirred solution of GG-4a (2.5 g, 8.6 mmol, 1.0 eq) in DCM (10 vol) at 0 ℃ was added Et3N (3.65 mL, 25.9 mmol, 3.0 eq), followed by methane sulfonyl chloride (1.32 mL, 17.2 mmol, 2.0 eq). The reaction mixture was warmed to room temperature and stirred at that temperature for 2 hours. An aqueous solution of NaCl (20 mL) was added to quench the reaction and the layers were separated. The organic layer was dried over anhydrous Na2SO4, the solids were removed by filtration, and the filtrate was concentrated in vacuo. Crude (2-((tert-butoxycarbonyl)amino)-3-(1H-indol-7-yl) propyl methanesulfonate), Compound GG- 5a (3 g, crude) was isolated as a pale yellow oil and carried forward without further purification. ESI-MS m/z: 369.2 [M+H] +. [0144] Step-4a: Synthesis of Compound GG-6a (tert-butyl (5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin- 5-yl) carbamate)):
[0143] To a stirred solution of GG-4a (2.5 g, 8.6 mmol, 1.0 eq) in DCM (10 vol) at 0 ℃ was added Et3N (3.65 mL, 25.9 mmol, 3.0 eq), followed by methane sulfonyl chloride (1.32 mL, 17.2 mmol, 2.0 eq). The reaction mixture was warmed to room temperature and stirred at that temperature for 2 hours. An aqueous solution of NaCl (20 mL) was added to quench the reaction and the layers were separated. The organic layer was dried over anhydrous Na2SO4, the solids were removed by filtration, and the filtrate was concentrated in vacuo. Crude (2-((tert-butoxycarbonyl)amino)-3-(1H-indol-7-yl) propyl methanesulfonate), Compound GG- 5a (3 g, crude) was isolated as a pale yellow oil and carried forward without further purification. ESI-MS m/z: 369.2 [M+H] +. [0144] Step-4a: Synthesis of Compound GG-6a (tert-butyl (5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin- 5-yl) carbamate)):
 [0145] To a stirred solution of Compound GG-5a (3 g crude, 8.14 mmol, 1.0 eq) in DMF (10 vol) at 0 ℃ was added NaH (0.78 g, 32.6 mmol, 4.0 eq). The reaction mixture was warmed to room temperature and stirred at that temperature for 2 hours. The reaction mixture was quenched with saturated aqueous NH4Cl solution (10 mL) and extracted with ethyl acetate (2 x 20 mL). The combined organic layers were dried over anhydrous Na2SO4, the solids were removed by filtration, and the filtrate was concentrated in vacuo. The crude material was purified by silica gel chromatography (20% EtOAc/Heptane) to afford (tert-butyl (5,6- dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5-yl) carbamate)), Compound GG-6a (1.3 g, 59% yield) as a white solid. ESI-MS m/z: 273.2 [M+H] +. [0146] Synthesis of Intermediate GG-6b: tert-butyl (9-chloro-8-fluoro-5,6-dihydro-4H-pyrrolo[3,2,1- ij]quinolin-5-yl)carbamate
WSGR Docket No.55776-726.601
[0145] To a stirred solution of Compound GG-5a (3 g crude, 8.14 mmol, 1.0 eq) in DMF (10 vol) at 0 ℃ was added NaH (0.78 g, 32.6 mmol, 4.0 eq). The reaction mixture was warmed to room temperature and stirred at that temperature for 2 hours. The reaction mixture was quenched with saturated aqueous NH4Cl solution (10 mL) and extracted with ethyl acetate (2 x 20 mL). The combined organic layers were dried over anhydrous Na2SO4, the solids were removed by filtration, and the filtrate was concentrated in vacuo. The crude material was purified by silica gel chromatography (20% EtOAc/Heptane) to afford (tert-butyl (5,6- dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5-yl) carbamate)), Compound GG-6a (1.3 g, 59% yield) as a white solid. ESI-MS m/z: 273.2 [M+H] +. [0146] Synthesis of Intermediate GG-6b: tert-butyl (9-chloro-8-fluoro-5,6-dihydro-4H-pyrrolo[3,2,1- ij]quinolin-5-yl)carbamate
WSGR Docket No.55776-726.601
 [0147] Step-1b: Synthesis of Int-3b amino)-3-(4-chloro-5-fluor o-1H- indol-7-yl)propanoatebutanoate):
[0147] Step-1b: Synthesis of Int-3b amino)-3-(4-chloro-5-fluor o-1H- indol-7-yl)propanoatebutanoate):
 [0152] To a stirred solution of Compound GG-4b (1.6 g, 4.67 mmol, 1.0 eq) in DCM (16 mL) at 0 ℃ was added Et3N (1.36 mL, 9.35 mmol, 2.0 eq), followed by methane sulfonyl chloride (0.43 mL, 5.60 mmol 1.2 eq). The reaction mixture was warmed to room temperature and stirred at that temperature for 1 hour. The reaction mixture was quenched with saturated aqueous NaHCO3 solution (10 mL) and extracted with DCM (2 x 10 mL). The combined organic layers were washed with an aqueous solution of NaCl (10 mL) and dried over anhydrous Na2SO4. The solids were removed by filtration and the filtrate was concentrated in vacuo to afford crude (2-((tert-butoxycarbonyl) amino)-3-(4-chloro-5-fluoro-1H-indol-7-yl) propyl methanesulfonate), Compound GG-5b (2 g, crude) as a yellow oil, which was carried forward without further purification. ESI-MS m/z: 421.2 [M+H] +. [0153] Step-4b: Synthesis of Compound GG-6b (tert-butyl (9-chloro-8-fluoro-5, 6-dihydro-4H- pyrrolo[3,2,1-ij]quinolin-5-yl)carbamate):
[0152] To a stirred solution of Compound GG-4b (1.6 g, 4.67 mmol, 1.0 eq) in DCM (16 mL) at 0 ℃ was added Et3N (1.36 mL, 9.35 mmol, 2.0 eq), followed by methane sulfonyl chloride (0.43 mL, 5.60 mmol 1.2 eq). The reaction mixture was warmed to room temperature and stirred at that temperature for 1 hour. The reaction mixture was quenched with saturated aqueous NaHCO3 solution (10 mL) and extracted with DCM (2 x 10 mL). The combined organic layers were washed with an aqueous solution of NaCl (10 mL) and dried over anhydrous Na2SO4. The solids were removed by filtration and the filtrate was concentrated in vacuo to afford crude (2-((tert-butoxycarbonyl) amino)-3-(4-chloro-5-fluoro-1H-indol-7-yl) propyl methanesulfonate), Compound GG-5b (2 g, crude) as a yellow oil, which was carried forward without further purification. ESI-MS m/z: 421.2 [M+H] +. [0153] Step-4b: Synthesis of Compound GG-6b (tert-butyl (9-chloro-8-fluoro-5, 6-dihydro-4H- pyrrolo[3,2,1-ij]quinolin-5-yl)carbamate):
 [0154] To a stirred solution of Compound GG-5b (1.0 g crude, 2.38 mmol, 1.0 eq) in DMF (10 vol) at 0 ℃ was added NaH (228 mg, 9.50 mmol, 4.0 eq) and the reaction mixture was stirred at 0 ℃ for 2 hours. The reaction mixture was quenched with saturated aqueous NH4Cl solution (10 mL) and extracted with ethyl acetate (2 x 20 mL). The combined organic layers were washed with an aqueous solution of NaCl (15 mL), the organic layer was dried over anhydrous Na2SO4, solids were removed by filtration, and the filtrate was concentrated in vacuo. The crude was purified by silica gel chromatography (20% EtOAc/Heptane), to afford (tert-butyl (9-chloro-8-fluoro-5, 6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5-yl)-carbamate), Compound GG-6b (0.3 g, 39% yield) as a yellow solid. ESI-MS m/z: 324.8 [M+H] +.
WSGR Docket No.55776-726.601 [0155] Synthesis of Intermediate Compound GG-6c: tert-butyl (8-fluoro-5,6-dihydro-4H- pyrrolo[3,2,1-ij]quinolin-5-yl)carbamate
[0154] To a stirred solution of Compound GG-5b (1.0 g crude, 2.38 mmol, 1.0 eq) in DMF (10 vol) at 0 ℃ was added NaH (228 mg, 9.50 mmol, 4.0 eq) and the reaction mixture was stirred at 0 ℃ for 2 hours. The reaction mixture was quenched with saturated aqueous NH4Cl solution (10 mL) and extracted with ethyl acetate (2 x 20 mL). The combined organic layers were washed with an aqueous solution of NaCl (15 mL), the organic layer was dried over anhydrous Na2SO4, solids were removed by filtration, and the filtrate was concentrated in vacuo. The crude was purified by silica gel chromatography (20% EtOAc/Heptane), to afford (tert-butyl (9-chloro-8-fluoro-5, 6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5-yl)-carbamate), Compound GG-6b (0.3 g, 39% yield) as a yellow solid. ESI-MS m/z: 324.8 [M+H] +.
WSGR Docket No.55776-726.601 [0155] Synthesis of Intermediate Compound GG-6c: tert-butyl (8-fluoro-5,6-dihydro-4H- pyrrolo[3,2,1-ij]quinolin-5-yl)carbamate
 [0156] Step-1c: Synthesis of Compound GG-3c (methyl 2-((tert-butoxycarbonyl)amino)-3-(5-fluoro- 1H-indol-7-yl)propanoate)):
[0156] Step-1c: Synthesis of Compound GG-3c (methyl 2-((tert-butoxycarbonyl)amino)-3-(5-fluoro- 1H-indol-7-yl)propanoate)):
 [0161] To a solution of Compound GG-4c (2.1 g, 6.81 mmol, 1.0 eq), in DCM (10 vol) at 0 ℃ was added Et3N (2.96 mL, 20.4 mmol, 3.0 eq), followed by methane sulfonyl chloride (11.0 mL, 13.6 mmol, 2.0 eq). Then the reaction mixture was stirred at room temperature for 2 hours. The reaction mixture was quenched with a saturated aqueous NaCl solution (20 mL), the layers were separated, and the organic phase was dried over anhydrous Na2SO4. The solids were removed by filtration and the filtrate was concentrated in vacuo, to give (2-((tert-butoxycarbonyl) amino)-3-(5-fluoro-1H-indol-7-yl)propylmehanesulfonate), Compound GG- 5c (2.8 g, crude) as a pale yellow oil, which was carried forward without further purification. ESI-MS m/z: 386.8 [M+H] +. [0162] Step-4c: Synthesis of Compound GG-6c: tert-butyl (8-fluoro-5,6-dihydro-4H-pyrrolo[3,2,1- ij]quinolin-5-yl)carbamate
[0161] To a solution of Compound GG-4c (2.1 g, 6.81 mmol, 1.0 eq), in DCM (10 vol) at 0 ℃ was added Et3N (2.96 mL, 20.4 mmol, 3.0 eq), followed by methane sulfonyl chloride (11.0 mL, 13.6 mmol, 2.0 eq). Then the reaction mixture was stirred at room temperature for 2 hours. The reaction mixture was quenched with a saturated aqueous NaCl solution (20 mL), the layers were separated, and the organic phase was dried over anhydrous Na2SO4. The solids were removed by filtration and the filtrate was concentrated in vacuo, to give (2-((tert-butoxycarbonyl) amino)-3-(5-fluoro-1H-indol-7-yl)propylmehanesulfonate), Compound GG- 5c (2.8 g, crude) as a pale yellow oil, which was carried forward without further purification. ESI-MS m/z: 386.8 [M+H] +. [0162] Step-4c: Synthesis of Compound GG-6c: tert-butyl (8-fluoro-5,6-dihydro-4H-pyrrolo[3,2,1- ij]quinolin-5-yl)carbamate
 [0163] To a solution of Compound GG-5c (2.8 g crude, 7.24 mmol, 1.0 eq) in DMF (28 mL) at 0 ℃ was added NaH (0.69 g, 29.0 mmol, 4.0 eq). The resulting reaction mixture was warmed to room temperature and stirred at that temperature for 2 hours. The reaction mixture was quenched with saturated NH4Cl solution (15 mL) and extracted with ethyl acetate (2 x 20 mL). The combined organic phase was dried over anhydrous Na2SO4, the solids were removed by filtration, and the filtrate was concentrated in vacuo. The crude material was purified by silica gel chromatography (20 % EtOAc/Heptane), to afford tert-butyl (8- fluoro-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5-yl)carbamate, Compound GG-6c (1.4 g, 67% yield) as an off white solid. ESI-MS m/z: 290.9 [M+H] +.
WSGR Docket No.55776-726.601 [0164] Synthesis of Intermediate Compound GG-6d: tert-butyl (8-chloro-9-fluoro-5,6-dihydro-4H- pyrrolo[3,2,1-ij]quinolin-5-yl)carbamate):
[0163] To a solution of Compound GG-5c (2.8 g crude, 7.24 mmol, 1.0 eq) in DMF (28 mL) at 0 ℃ was added NaH (0.69 g, 29.0 mmol, 4.0 eq). The resulting reaction mixture was warmed to room temperature and stirred at that temperature for 2 hours. The reaction mixture was quenched with saturated NH4Cl solution (15 mL) and extracted with ethyl acetate (2 x 20 mL). The combined organic phase was dried over anhydrous Na2SO4, the solids were removed by filtration, and the filtrate was concentrated in vacuo. The crude material was purified by silica gel chromatography (20 % EtOAc/Heptane), to afford tert-butyl (8- fluoro-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5-yl)carbamate, Compound GG-6c (1.4 g, 67% yield) as an off white solid. ESI-MS m/z: 290.9 [M+H] +.
WSGR Docket No.55776-726.601 [0164] Synthesis of Intermediate Compound GG-6d: tert-butyl (8-chloro-9-fluoro-5,6-dihydro-4H- pyrrolo[3,2,1-ij]quinolin-5-yl)carbamate):
 [0165] Step-1d: Synthesis of Compound GG-3d (methyl 2-((tert-butoxycarbonyl)amino)-3-(5-chloro- 4-fluor o-1H-indol-7-yl)propanoate):
[0165] Step-1d: Synthesis of Compound GG-3d (methyl 2-((tert-butoxycarbonyl)amino)-3-(5-chloro- 4-fluor o-1H-indol-7-yl)propanoate):
 [0170] To a solution of Compound GG-4d (1.6 g, 4.67 mmol, 1.0 eq), in DCM (16 mL) at 0 ℃ was added Et3N (1.36 mL, 9.32 mmol, 2.0 eq), followed by methanesulfonyl chloride (0.43 mL, 5.60 mmol 1.2 eq). The reaction mixture was warmed to room temperature and stirred at that temperature for 1 hour. The reaction mixture was quenched with a saturated aqueous NaHCO3 solution (20 mL) and extracted with DCM (2 x 20 mL). The combined organic layers were washed with an aqueous solution of NaCl (20 mL) and dried over anhydrous Na2SO4. The solids were removed by filtration and the filtrate was concentrated in vacuo to give crude 2-((tert-butoxycarbonyl)amino)-3-(5-chloro-4-fluoro-1H-In dol-7-yl)propyl methanesulfonate), Compound GG-5d (2 g, crude) as a yellow oil, which was carried forward without further purification. ESI- MS m/z: 421.9. [M+H]+. [0171] Step-4d; Synthesis of Compound GG-6d: tert-butyl (8-chloro-9-fluoro-5,6-dihydro-4H- pyrrolo[3,2,1-ij]quinolin-5-yl)carbamate):
[0170] To a solution of Compound GG-4d (1.6 g, 4.67 mmol, 1.0 eq), in DCM (16 mL) at 0 ℃ was added Et3N (1.36 mL, 9.32 mmol, 2.0 eq), followed by methanesulfonyl chloride (0.43 mL, 5.60 mmol 1.2 eq). The reaction mixture was warmed to room temperature and stirred at that temperature for 1 hour. The reaction mixture was quenched with a saturated aqueous NaHCO3 solution (20 mL) and extracted with DCM (2 x 20 mL). The combined organic layers were washed with an aqueous solution of NaCl (20 mL) and dried over anhydrous Na2SO4. The solids were removed by filtration and the filtrate was concentrated in vacuo to give crude 2-((tert-butoxycarbonyl)amino)-3-(5-chloro-4-fluoro-1H-In dol-7-yl)propyl methanesulfonate), Compound GG-5d (2 g, crude) as a yellow oil, which was carried forward without further purification. ESI- MS m/z: 421.9. [M+H]+. [0171] Step-4d; Synthesis of Compound GG-6d: tert-butyl (8-chloro-9-fluoro-5,6-dihydro-4H- pyrrolo[3,2,1-ij]quinolin-5-yl)carbamate):
 [0175] Step-1: Synthesis of Compound GG-9: (tert-butyl (8-fluoro-9-methyl-5,6-dihydro-4H- pyrrolo[3,2,1-ij]quiolin-5-yl)carbamate):
[0175] Step-1: Synthesis of Compound GG-9: (tert-butyl (8-fluoro-9-methyl-5,6-dihydro-4H- pyrrolo[3,2,1-ij]quiolin-5-yl)carbamate):

 [0176] To a stirred solution of Compound GG-6b (50 mg, 0.15 mmol, 1.0 eq) in dioxane (5 mL) was added methylboronic acid (18 mg, 0.30 mmol, 2.0 eq) and KOAc (29.4 mg, 0.30 mmol, 2.0 eq) at room temperature. Nitrogen was bubbled through the reaction mixture for 10 minutes, then X-Phos (14.2 mg, 0.03 mmol, 0.2 eq) was added, followed by Pd2(dba)3 (13.74 mg, 0.015 mmol 0.1 eq). Nitrogen was again bubbled through the reaction mixture for 5 minutes, and then the reaction mixture was heated to 80 ℃ and stirred at that temperature for 6 hours. The reaction mixture was cooled to room temperature, quenched with a saturated aqueous NH4Cl solution (5 mL), and extracted with DCM (2 x 5 mL). The combined organic layers were washed with an aqueous solution of NaCl (10 mL), the organic layer was dried over anhydrous Na2SO4, the solids were filtered, and the filtrate was concentrated in vacuo. The crude material was purified by silica gel chromatography (18% EtOAc/Heptane) to afford (tert-butyl (8-fluoro-9-methyl-5,6-dihydro- 4H-pyrrolo[3,2,1-ij]quiolin-5-yl)carbamate), Compound GG-9 (30 mg, 64% yield) as a pale yellow film. ESI-MS m/z: 304.8 [M+H]+. [0177] Step-2: Synthesis of 8-fluoro-N,9-dimethyl-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5-amine; Compound 67
WSGR Docket No.55776-726.601
[0176] To a stirred solution of Compound GG-6b (50 mg, 0.15 mmol, 1.0 eq) in dioxane (5 mL) was added methylboronic acid (18 mg, 0.30 mmol, 2.0 eq) and KOAc (29.4 mg, 0.30 mmol, 2.0 eq) at room temperature. Nitrogen was bubbled through the reaction mixture for 10 minutes, then X-Phos (14.2 mg, 0.03 mmol, 0.2 eq) was added, followed by Pd2(dba)3 (13.74 mg, 0.015 mmol 0.1 eq). Nitrogen was again bubbled through the reaction mixture for 5 minutes, and then the reaction mixture was heated to 80 ℃ and stirred at that temperature for 6 hours. The reaction mixture was cooled to room temperature, quenched with a saturated aqueous NH4Cl solution (5 mL), and extracted with DCM (2 x 5 mL). The combined organic layers were washed with an aqueous solution of NaCl (10 mL), the organic layer was dried over anhydrous Na2SO4, the solids were filtered, and the filtrate was concentrated in vacuo. The crude material was purified by silica gel chromatography (18% EtOAc/Heptane) to afford (tert-butyl (8-fluoro-9-methyl-5,6-dihydro- 4H-pyrrolo[3,2,1-ij]quiolin-5-yl)carbamate), Compound GG-9 (30 mg, 64% yield) as a pale yellow film. ESI-MS m/z: 304.8 [M+H]+. [0177] Step-2: Synthesis of 8-fluoro-N,9-dimethyl-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5-amine; Compound 67
WSGR Docket No.55776-726.601
 To a solution of mg, 0 ℃ was added a LiAlH4 solution (2M in THF, 0.19 mL, 0.39 mmol, 4.0 eq). The reaction mixture was heated to reflux and stirred at that temperature for 2 hours. The reaction mixture was cooled to 0 ℃, quenched with a saturated aqueous NH4Cl solution (5 mL), and extracted with DCM (2 x 5 mL). The combined organic layers were washed with an aqueous solution of NaCl (10 mL), the organic layer was dried over anhydrous Na2SO4, the solids were filtered, and the filtrate was concentrated in vacuo. The crude material was submitted for RP prep HPLC purification to afford 8-fluoro-N,9-dimethyl-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5-amine, Compound 67 (15 mg, 70% yield). ESI-MS m/z: 219.1 [M+H] +.1H NMR (400 MHz, DMSO-d6) δ: 7.32 (d, J = 2.9 Hz, 1H), 6.69 (d, J = 10.5 Hz, 1H), 6.39 (d, J = 3.1 Hz, 1H), 4.32 (dd, J = 3.7, 12.0 Hz, 1H), 3.86 - 3.80 (m, 1H), 3.17 - 3.09 (m, 2H), 2.79 - 2.73 (m, 1H), 2.39 (s, 3H), 2.31 (d, J = 1.3 Hz, 3H). Example A4: [0178] Synthesis of 8-fluoro-N,N,9-trimethyl-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5-amine; Compound 73 [0179] Scheme-4: Synthesis of 8-fluoro-N,N,9-trimethyl-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5- amine; Compound 73:
To a solution of mg, 0 ℃ was added a LiAlH4 solution (2M in THF, 0.19 mL, 0.39 mmol, 4.0 eq). The reaction mixture was heated to reflux and stirred at that temperature for 2 hours. The reaction mixture was cooled to 0 ℃, quenched with a saturated aqueous NH4Cl solution (5 mL), and extracted with DCM (2 x 5 mL). The combined organic layers were washed with an aqueous solution of NaCl (10 mL), the organic layer was dried over anhydrous Na2SO4, the solids were filtered, and the filtrate was concentrated in vacuo. The crude material was submitted for RP prep HPLC purification to afford 8-fluoro-N,9-dimethyl-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5-amine, Compound 67 (15 mg, 70% yield). ESI-MS m/z: 219.1 [M+H] +.1H NMR (400 MHz, DMSO-d6) δ: 7.32 (d, J = 2.9 Hz, 1H), 6.69 (d, J = 10.5 Hz, 1H), 6.39 (d, J = 3.1 Hz, 1H), 4.32 (dd, J = 3.7, 12.0 Hz, 1H), 3.86 - 3.80 (m, 1H), 3.17 - 3.09 (m, 2H), 2.79 - 2.73 (m, 1H), 2.39 (s, 3H), 2.31 (d, J = 1.3 Hz, 3H). Example A4: [0178] Synthesis of 8-fluoro-N,N,9-trimethyl-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5-amine; Compound 73 [0179] Scheme-4: Synthesis of 8-fluoro-N,N,9-trimethyl-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5- amine; Compound 73:
 [0180] Step-1: Synthesis of GG-10:(tert-butyl (8-fluoro-9-methyl-5,6-dihydro-4H-pyrrolo[3,2,1-ij] quinolin-5-yl)(methyl)
[0180] Step-1: Synthesis of GG-10:(tert-butyl (8-fluoro-9-methyl-5,6-dihydro-4H-pyrrolo[3,2,1-ij] quinolin-5-yl)(methyl)
 WSGR Docket No.55776-726.601 [0181] To a solution of Compound GG-9 (80 mg, 0.26 mmol, 1.0 eq) in DMF (10 vol) at 0 ℃ was added NaH (14.4 mg, 3.94 mmol, 1.5 eq) and the reaction mixture was stirred for 10 min. MeI (0.03 mL, 0.52 mmol, 2.0 eq) was added at 0 ℃ and the resulting mixture was warmed to room temperature and stirred at that temperature for 2 hours. The reaction mixture was diluted with ice cold water (5 mL) and extracted with ethyl acetate (2 x 5 mL). The combined organic layers were washed with an aqueous solution of NaCl (10 mL), the organic layer was dried over anhydrous Na2SO4, the solids were removed by filtration, and the filtrate was concentrated in vacuo to afford (tert-butyl (8-fluoro-9-methyl-5,6-dihydro-4H-pyrrolo[3,2,1-ij] quinolin-5-yl)(methyl)carbamate), Compound GG-10 (80 mg, crude) as a pale yellow film. The crude material was carried forward without further purification. ESI-MS m/z: 319.30 [M+H] +. [0182] Step-2: Synthesis of 8-fluoro-N,N,9-trimethyl-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5- amine; Compound 73:
WSGR Docket No.55776-726.601 [0181] To a solution of Compound GG-9 (80 mg, 0.26 mmol, 1.0 eq) in DMF (10 vol) at 0 ℃ was added NaH (14.4 mg, 3.94 mmol, 1.5 eq) and the reaction mixture was stirred for 10 min. MeI (0.03 mL, 0.52 mmol, 2.0 eq) was added at 0 ℃ and the resulting mixture was warmed to room temperature and stirred at that temperature for 2 hours. The reaction mixture was diluted with ice cold water (5 mL) and extracted with ethyl acetate (2 x 5 mL). The combined organic layers were washed with an aqueous solution of NaCl (10 mL), the organic layer was dried over anhydrous Na2SO4, the solids were removed by filtration, and the filtrate was concentrated in vacuo to afford (tert-butyl (8-fluoro-9-methyl-5,6-dihydro-4H-pyrrolo[3,2,1-ij] quinolin-5-yl)(methyl)carbamate), Compound GG-10 (80 mg, crude) as a pale yellow film. The crude material was carried forward without further purification. ESI-MS m/z: 319.30 [M+H] +. [0182] Step-2: Synthesis of 8-fluoro-N,N,9-trimethyl-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5- amine; Compound 73:
 [0183] To a solution of Compound GG-10 (80 mg, 0.25 mmol, 1.0 eq) in DCM (10 vol) at 0 ℃ was added a LiAlH4 solution (2M in THF, 0.50 mL, 1.0 mmol, 4.0 eq). The reaction mixture was heated to reflux and stirred at that temperature for 2 hours. The reaction mixture was cooled to 0 ℃, quenched with a saturated aqueous NH4Cl solution (5 mL), and extracted with DCM (2 x 5 mL). The combined organic layers were washed with an aqueous solution of NaCl (10 mL), the organic layer was dried over anhydrous Na2SO4, the solids were filtered, and the filtrate was concentrated in vacuo. The crude material was purified by RP prep HPLC purification to give 8-fluoro-N,N,9-trimethyl-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5-amine, Compound 73 (30 mg, 52% yield). ESI-MS m/z: 233.15 [M+H] +.1H NMR (400 MHz, DMSO-d6) δ: 7.31 - 7.28 (m, 1H), 6.72 - 6.66 (m, 1H), 6.37 (d, J = 3.1 Hz, 1H), 4.34 - 4.25 (m, 1H), 4.02 - 3.95 (m, 1H), 3.05 - 2.87 (m, 3H), 2.31 - 2.29 (m, 9H) ppm. Prep HPLC purification details: COLUMN x-select C18 (250 * 30mm); 5µm Mobile Phase A 10 mM Ammonium bicarbonate in H2O Mobile Phase B 100% ACN Flow rate 25 mL/min Instrument ID Prep-14 Gradient (Time/%B) 0/10, 3/10, 18/95, 20/95, 22/10
WSGR Docket No.55776-726.601 Example A5: [0184] Synthesis of (R)-9-chloro-8-fluoro-N-methyl-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5-amine (Compound 62) and (S)-9-chloro-8-fluoro-N-methyl-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5-amine (Compound 61) [0185] Scheme-5: Synthesis of (R)-9-chloro-8-fluoro-N-methyl-5,6-dihydro-4H-pyrrolo[3,2,1- ij]quinolin-5-amine (Compound 62) and (S)-9-chloro-8-fluoro-N-methyl-5,6-dihydro-4H- pyrrolo[3,2,1-ij]quinolin-5-amine (Compound 61):
[0183] To a solution of Compound GG-10 (80 mg, 0.25 mmol, 1.0 eq) in DCM (10 vol) at 0 ℃ was added a LiAlH4 solution (2M in THF, 0.50 mL, 1.0 mmol, 4.0 eq). The reaction mixture was heated to reflux and stirred at that temperature for 2 hours. The reaction mixture was cooled to 0 ℃, quenched with a saturated aqueous NH4Cl solution (5 mL), and extracted with DCM (2 x 5 mL). The combined organic layers were washed with an aqueous solution of NaCl (10 mL), the organic layer was dried over anhydrous Na2SO4, the solids were filtered, and the filtrate was concentrated in vacuo. The crude material was purified by RP prep HPLC purification to give 8-fluoro-N,N,9-trimethyl-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5-amine, Compound 73 (30 mg, 52% yield). ESI-MS m/z: 233.15 [M+H] +.1H NMR (400 MHz, DMSO-d6) δ: 7.31 - 7.28 (m, 1H), 6.72 - 6.66 (m, 1H), 6.37 (d, J = 3.1 Hz, 1H), 4.34 - 4.25 (m, 1H), 4.02 - 3.95 (m, 1H), 3.05 - 2.87 (m, 3H), 2.31 - 2.29 (m, 9H) ppm. Prep HPLC purification details: COLUMN x-select C18 (250 * 30mm); 5µm Mobile Phase A 10 mM Ammonium bicarbonate in H2O Mobile Phase B 100% ACN Flow rate 25 mL/min Instrument ID Prep-14 Gradient (Time/%B) 0/10, 3/10, 18/95, 20/95, 22/10
WSGR Docket No.55776-726.601 Example A5: [0184] Synthesis of (R)-9-chloro-8-fluoro-N-methyl-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5-amine (Compound 62) and (S)-9-chloro-8-fluoro-N-methyl-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5-amine (Compound 61) [0185] Scheme-5: Synthesis of (R)-9-chloro-8-fluoro-N-methyl-5,6-dihydro-4H-pyrrolo[3,2,1- ij]quinolin-5-amine (Compound 62) and (S)-9-chloro-8-fluoro-N-methyl-5,6-dihydro-4H- pyrrolo[3,2,1-ij]quinolin-5-amine (Compound 61):
 [0186] Step-1: Chiral Resolution of (tert-butyl (9-chloro-8-fluoro-5, 6-dihydro-4H-pyrrolo[3,2,1- ij]quinolin-5-yl)-carbamate), Compound GG-6b: [0187] 250 mg of Compound GG-6b was submitted to chiral resolution (details below) The faster eluting peak was assigned as Compound GG-6b-Peak1 (60 mg, yellow solid, ESI-MS m/z: 304.8 [M+H]+), and the more slowly eluting peak was assigned as Compound GG-6b-Peak2 (60 mg, yellow solid, ESI-MS m/z: 304.8 [M+H]+). Chiral HPLC purification details: COLUMN Amylose-2 (30 x 250 mm, 5μm) Mobile Phase A 0.1% Isopropylamine in n- Hexane Mobile Phase B Isopropylamine Eluent A:B 95:05 Flow rate 50 mL/min Diluent Isopropanol + EtOH Detection wavelength 280 nm [0188] Step-2a: Synthesis of (R)-9-chloro-8-fluoro-N-methyl-5,6-dihydro-4H-pyrrolo[3,2,1- ij]quinolin-5-amine;
[0186] Step-1: Chiral Resolution of (tert-butyl (9-chloro-8-fluoro-5, 6-dihydro-4H-pyrrolo[3,2,1- ij]quinolin-5-yl)-carbamate), Compound GG-6b: [0187] 250 mg of Compound GG-6b was submitted to chiral resolution (details below) The faster eluting peak was assigned as Compound GG-6b-Peak1 (60 mg, yellow solid, ESI-MS m/z: 304.8 [M+H]+), and the more slowly eluting peak was assigned as Compound GG-6b-Peak2 (60 mg, yellow solid, ESI-MS m/z: 304.8 [M+H]+). Chiral HPLC purification details: COLUMN Amylose-2 (30 x 250 mm, 5μm) Mobile Phase A 0.1% Isopropylamine in n- Hexane Mobile Phase B Isopropylamine Eluent A:B 95:05 Flow rate 50 mL/min Diluent Isopropanol + EtOH Detection wavelength 280 nm [0188] Step-2a: Synthesis of (R)-9-chloro-8-fluoro-N-methyl-5,6-dihydro-4H-pyrrolo[3,2,1- ij]quinolin-5-amine;
 WSGR Docket No.55776-726.601 [0189] To a solution of Compound GG-6b-Peak1 (60 mg, 0.18 mmol, 1.0 eq) in DCM (10 vol) at 0 ℃ was added a LiAlH4 solution (2M in THF, 0.70 mL, 1.41 mmol, 4.0 eq). The reaction mixture was heated to reflux and was stirred at that temperature for 2 hours. The reaction mixture was cooled to 0 ℃, quenched with a saturated aqueous NH4Cl solution (5 mL), and extracted with DCM (2 x 5 mL). The combined organic layers were washed with an aqueous solution of NaCl, the organic layer was dried over anhydrous Na2SO4, the solids were filtered, and the filtrate was concentrated in vacuo. The crude material was purified by RP prep HPLC purification to afford (R)-9-chloro-8-fluoro-N-methyl-5,6-dihydro-4H-pyrrolo[3,2,1- ij]quinolin-5-amine, Compound 62 (15 mg, 34% yield). ESI-MS m/z: 239.1 [M+H] +.1H NMR (400 MHz, DMSO-d6) δ: 7.48 - 7.47 (m, 1H), 6.95 - 6.91 (m, 1H), 6.42 (d, J = 3.0 Hz, 1H), 4.33 (dd, J = 3.9, 11.9 Hz, 1H), 3.93 - 3.88 (m, 1H), 3.22 - 3.11 (m, 2H), 2.84 - 2.76 (m, 1H), 2.37 (s, 3H). Prep HPLC purification details: COLUMN x-select C18 (250 * 30mm); 5µm Mobile Phase A 10 mM Ammonium bicarbonate in H2O Mobile Phase B 100% ACN Flow rate 25 mL/min Instrument ID Prep-14 Gradient (Time/%B) 0/10, 3/10, 18/95, 20/95, 22/10 [0190] Step-2b: Synthesis of (S)-9-chloro-8-fluoro-N-methyl-5,6-dihydro-4H-pyrrolo[3,2,1- ij]quinolin-5-amine; Compound 61:
WSGR Docket No.55776-726.601 [0189] To a solution of Compound GG-6b-Peak1 (60 mg, 0.18 mmol, 1.0 eq) in DCM (10 vol) at 0 ℃ was added a LiAlH4 solution (2M in THF, 0.70 mL, 1.41 mmol, 4.0 eq). The reaction mixture was heated to reflux and was stirred at that temperature for 2 hours. The reaction mixture was cooled to 0 ℃, quenched with a saturated aqueous NH4Cl solution (5 mL), and extracted with DCM (2 x 5 mL). The combined organic layers were washed with an aqueous solution of NaCl, the organic layer was dried over anhydrous Na2SO4, the solids were filtered, and the filtrate was concentrated in vacuo. The crude material was purified by RP prep HPLC purification to afford (R)-9-chloro-8-fluoro-N-methyl-5,6-dihydro-4H-pyrrolo[3,2,1- ij]quinolin-5-amine, Compound 62 (15 mg, 34% yield). ESI-MS m/z: 239.1 [M+H] +.1H NMR (400 MHz, DMSO-d6) δ: 7.48 - 7.47 (m, 1H), 6.95 - 6.91 (m, 1H), 6.42 (d, J = 3.0 Hz, 1H), 4.33 (dd, J = 3.9, 11.9 Hz, 1H), 3.93 - 3.88 (m, 1H), 3.22 - 3.11 (m, 2H), 2.84 - 2.76 (m, 1H), 2.37 (s, 3H). Prep HPLC purification details: COLUMN x-select C18 (250 * 30mm); 5µm Mobile Phase A 10 mM Ammonium bicarbonate in H2O Mobile Phase B 100% ACN Flow rate 25 mL/min Instrument ID Prep-14 Gradient (Time/%B) 0/10, 3/10, 18/95, 20/95, 22/10 [0190] Step-2b: Synthesis of (S)-9-chloro-8-fluoro-N-methyl-5,6-dihydro-4H-pyrrolo[3,2,1- ij]quinolin-5-amine; Compound 61:
 [0191] To a solution of Compound GG-6b-Peak2 (60 mg, 0.18 mmol, 1.0 eq) in DCM (10 vol) at 0 ℃ was added a LiAlH4 solution (2M in THF, 0.70 mL, 1.41 mmol, 4.0 eq). The reaction mixture was heated to reflux and was stirred at that temperature for 2 hours. The reaction mixture was cooled to 0 ℃, quenched with a saturated aqueous NH4Cl solution (5 mL), and extracted with DCM (2 x 5 mL). The combined organic layers were washed with an aqueous solution of NaCl, the organic layer was dried over anhydrous Na2SO4, the solids were filtered, and the filtrate was concentrated in vacuo. The crude material was purified by RP prep HPLC purification to afford (S)-9-chloro-8-fluoro-N-methyl-5,6-dihydro-4H-pyrrolo[3,2,1- ij]quinolin-5-amine, Compound 61 (15 mg, 34% yield) . ESI-MS m/z: 239.1 [M+H] +.1H NMR (400 MHz, DMSO-d6) δ: 7.49 - 7.47 (m, 1H), 6.94 (d, J = 10.1 Hz, 1H), 6.42 (d, J = 3.0 Hz, 1H), 4.33 (dd, J = 3.8, 12.3 Hz, 1H), 3.92 (dd, J = 6.9, 12.3 Hz, 1H), 3.22 - 3.11 (m, 2H), 2.84 - 2.77 (m, 1H), 2.40 - 2.37 (m, 3H).
WSGR Docket No.55776-726.601 Prep HPLC purification details: COLUMN x-select C18 (250 * 30mm); 5 µm Mobile Phase A 10 mM Ammonium bicarbonate in H2O Mobile Phase B 100% ACN Flow rate 25 mL/min Instrument ID Prep-14 Gradient (Time/%B) 0/10, 3/10, 18/95, 20/95, 22/10 Example A6: [0192] Synthesis of 9-chloro-8-fluoro-N,N-dimethyl-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5- amine, Compound 71 [0193] Scheme-6: Synthesis of 9-chloro-8-fluoro-N,N-dimethyl-5,6-dihydro-4H-pyrrolo[3,2,1- ij]quinolin-5-amine, Compound 71
[0191] To a solution of Compound GG-6b-Peak2 (60 mg, 0.18 mmol, 1.0 eq) in DCM (10 vol) at 0 ℃ was added a LiAlH4 solution (2M in THF, 0.70 mL, 1.41 mmol, 4.0 eq). The reaction mixture was heated to reflux and was stirred at that temperature for 2 hours. The reaction mixture was cooled to 0 ℃, quenched with a saturated aqueous NH4Cl solution (5 mL), and extracted with DCM (2 x 5 mL). The combined organic layers were washed with an aqueous solution of NaCl, the organic layer was dried over anhydrous Na2SO4, the solids were filtered, and the filtrate was concentrated in vacuo. The crude material was purified by RP prep HPLC purification to afford (S)-9-chloro-8-fluoro-N-methyl-5,6-dihydro-4H-pyrrolo[3,2,1- ij]quinolin-5-amine, Compound 61 (15 mg, 34% yield) . ESI-MS m/z: 239.1 [M+H] +.1H NMR (400 MHz, DMSO-d6) δ: 7.49 - 7.47 (m, 1H), 6.94 (d, J = 10.1 Hz, 1H), 6.42 (d, J = 3.0 Hz, 1H), 4.33 (dd, J = 3.8, 12.3 Hz, 1H), 3.92 (dd, J = 6.9, 12.3 Hz, 1H), 3.22 - 3.11 (m, 2H), 2.84 - 2.77 (m, 1H), 2.40 - 2.37 (m, 3H).
WSGR Docket No.55776-726.601 Prep HPLC purification details: COLUMN x-select C18 (250 * 30mm); 5 µm Mobile Phase A 10 mM Ammonium bicarbonate in H2O Mobile Phase B 100% ACN Flow rate 25 mL/min Instrument ID Prep-14 Gradient (Time/%B) 0/10, 3/10, 18/95, 20/95, 22/10 Example A6: [0192] Synthesis of 9-chloro-8-fluoro-N,N-dimethyl-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5- amine, Compound 71 [0193] Scheme-6: Synthesis of 9-chloro-8-fluoro-N,N-dimethyl-5,6-dihydro-4H-pyrrolo[3,2,1- ij]quinolin-5-amine, Compound 71
 [0194] Step-1: Synthesis of Compound GG-11; (tert-butyl (9-chloro-8-fluoro-5,6-dihydro-4H- pyrrolo[3,2,1-ij] quinolin-5-yl)(methyl)carbamate):
[0194] Step-1: Synthesis of Compound GG-11; (tert-butyl (9-chloro-8-fluoro-5,6-dihydro-4H- pyrrolo[3,2,1-ij] quinolin-5-yl)(methyl)carbamate):
 [0195] To a solution of Compound GG-6b (100 mg, 0.31 mmol, 1.0 eq) in DMF (10 vol) at 0 ℃ was added NaH (14.8 mg, 0.61 mmol, 2.0 eq) and the reaction mixture was stirred at 0 ℃ for 10 minutes. MeI (0.04 mL, 0.61 mmol, 2.0 eq) was added at 0 ℃ and the resulting mixture was warmed to room temperature and stirred at that temperature for 30 min. The reaction mixture was diluted with ice cold water (5 mL) and extracted with ethyl acetate (2 x 5 mL). The combined organic layers were washed with an aqueous NaCl solution (5 mL), dried over anhydrous Na2SO4, the solids were filtered, and the filtrate was concentrated in vacuo. Crude (tert-butyl (9-chloro-8-fluoro-5,6-dihydro-4H-pyrrolo[3,2,1-ij] quinolin-5-yl)(methyl)-
WSGR Docket No.55776-726.601 carbamate), Compound GG-11 (100 mg, crude) was isolated as a yellow solid and carried forward without further purification. ESI-MS m/z: 338.9 [M+H] + [0196] Step-2: Synthesis of 9-chloro-8-fluoro-N,N-dimethyl-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin- 5-amine; Compound 71
[0195] To a solution of Compound GG-6b (100 mg, 0.31 mmol, 1.0 eq) in DMF (10 vol) at 0 ℃ was added NaH (14.8 mg, 0.61 mmol, 2.0 eq) and the reaction mixture was stirred at 0 ℃ for 10 minutes. MeI (0.04 mL, 0.61 mmol, 2.0 eq) was added at 0 ℃ and the resulting mixture was warmed to room temperature and stirred at that temperature for 30 min. The reaction mixture was diluted with ice cold water (5 mL) and extracted with ethyl acetate (2 x 5 mL). The combined organic layers were washed with an aqueous NaCl solution (5 mL), dried over anhydrous Na2SO4, the solids were filtered, and the filtrate was concentrated in vacuo. Crude (tert-butyl (9-chloro-8-fluoro-5,6-dihydro-4H-pyrrolo[3,2,1-ij] quinolin-5-yl)(methyl)-
WSGR Docket No.55776-726.601 carbamate), Compound GG-11 (100 mg, crude) was isolated as a yellow solid and carried forward without further purification. ESI-MS m/z: 338.9 [M+H] + [0196] Step-2: Synthesis of 9-chloro-8-fluoro-N,N-dimethyl-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin- 5-amine; Compound 71
 [0197] To a solution of Compound GG-11 (100 mg, 0.29 mmol, 1.0 eq) in DCM (10 vol) at 0 ℃ was added a LiAlH4 solution (2M in THF, 0.29 mL, 0.58 mmol, 2.0 eq). The resulting reaction mixture was heated to reflux and was stirred at that temperature for 2 hours. The reaction mixture was cooled to 0 ℃ and quenched with a saturated aqueous NH4Cl solution (5 mL). The formed solids were filtered off and washed with multiple portions of ethyl acetate. The combined organic extracts were washed with an aqueous solution of NaCl (10 mL), the organic layer was dried over anhydrous Na2SO4, the solids were filtered, and the filtrate was concentrated in vacuo. The crude material was purified by silica gel chromatography (3% MeOH/DCM) to afford 9-chloro-8-fluoro-N,N-dimethyl-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5-amine, Compound 71 (50 mg, 68% yield). ESI-MS m/z: 252.9 [M+H] +.1H NMR (400 MHz, DMSO-d6) δ: 7.49 - 7.46 (m, 1H), 6.95 (br d, J = 10.4 Hz, 1H), 6.42 (br d, J = 2.5 Hz, 1H), 4.38 - 4.32 (m, 1H), 4.12 - 4.07 (m, 1H), 3.13 - 2.99 (m, 3H), 2.30 (s, 6H). Example A7: [0198] Synthesis of 9-chloro-8-fluoro-N-methyl-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5-amine; Compound 68:
[0197] To a solution of Compound GG-11 (100 mg, 0.29 mmol, 1.0 eq) in DCM (10 vol) at 0 ℃ was added a LiAlH4 solution (2M in THF, 0.29 mL, 0.58 mmol, 2.0 eq). The resulting reaction mixture was heated to reflux and was stirred at that temperature for 2 hours. The reaction mixture was cooled to 0 ℃ and quenched with a saturated aqueous NH4Cl solution (5 mL). The formed solids were filtered off and washed with multiple portions of ethyl acetate. The combined organic extracts were washed with an aqueous solution of NaCl (10 mL), the organic layer was dried over anhydrous Na2SO4, the solids were filtered, and the filtrate was concentrated in vacuo. The crude material was purified by silica gel chromatography (3% MeOH/DCM) to afford 9-chloro-8-fluoro-N,N-dimethyl-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5-amine, Compound 71 (50 mg, 68% yield). ESI-MS m/z: 252.9 [M+H] +.1H NMR (400 MHz, DMSO-d6) δ: 7.49 - 7.46 (m, 1H), 6.95 (br d, J = 10.4 Hz, 1H), 6.42 (br d, J = 2.5 Hz, 1H), 4.38 - 4.32 (m, 1H), 4.12 - 4.07 (m, 1H), 3.13 - 2.99 (m, 3H), 2.30 (s, 6H). Example A7: [0198] Synthesis of 9-chloro-8-fluoro-N-methyl-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5-amine; Compound 68:
 [0199] To a solution of Compound GG-6b (40 mg, 0.12 mmol, 1.0 eq) in DCM (10 vol) at 0 ℃ was added a LiAlH4 solution (2M in THF, 0.15 mL, 0.30 mmol, 2.5 eq). The resulting reaction mixture was heated to reflux and was stirred at that temperature for 2 hours. The reaction mixture was cooled to 0 ℃ and quenched with a saturated aqueous NH4Cl solution (5 mL). The formed solids were filtered off and washed with multiple portions of ethyl acetate. The combined organic extracts were washed with an aqueous solution of
WSGR Docket No.55776-726.601 NaCl (10 mL), the organic layer was dried over anhydrous Na2SO4, the solids were filtered, and the filtrate was concentrated in vacuo. The crude was purified by silica gel chromatography (3% MeOH/DCM) to afford 9-chloro-8-fluoro-N-methyl-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5-amine, Compound 68 (20 mg, 70% yield). ESI-MS m/z: 239.3 [M+H] +.1H NMR (400 MHz, DMSO-d6) δ: 7.47 (br d, J = 2.1 Hz, 1H), 6.93 (br d, J = 10.4 Hz, 1H), 6.41 (br d, J = 2.1 Hz, 1H), 4.32 (br dd, J = 2.7, 12.2 Hz, 1H), 3.89 (br dd, J = 7.0, 12.0 Hz, 1H), 3.13 (br d, J = 13.3 Hz, 2H), 2.82 - 2.75 (m, 1H), 2.36 (s, 3H). Example A8: [0200] Synthesis of (R)-9-chloro-8-fluoro-N,N-dimethyl-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5- amine (Compound 65) and (S)-9-chloro-8-fluoro-N,N-dimethyl-5,6-dihydro-4H-pyrrolo[3,2,1- ij]quinolin-5-amine (Compound 64) [0201] Scheme-7: Synthesis of (R)-9-chloro-8-fluoro-N,N-dimethyl-5,6-dihydro-4H-pyrrolo[3,2,1- ij]quinolin-5-amine (Compound 65) and (S)-9-chloro-8-fluoro-N,N-dimethyl-5,6-dihydro-4H- pyrrolo[3,2,1-ij]quinolin-5-amine (Compound 64)
[0199] To a solution of Compound GG-6b (40 mg, 0.12 mmol, 1.0 eq) in DCM (10 vol) at 0 ℃ was added a LiAlH4 solution (2M in THF, 0.15 mL, 0.30 mmol, 2.5 eq). The resulting reaction mixture was heated to reflux and was stirred at that temperature for 2 hours. The reaction mixture was cooled to 0 ℃ and quenched with a saturated aqueous NH4Cl solution (5 mL). The formed solids were filtered off and washed with multiple portions of ethyl acetate. The combined organic extracts were washed with an aqueous solution of
WSGR Docket No.55776-726.601 NaCl (10 mL), the organic layer was dried over anhydrous Na2SO4, the solids were filtered, and the filtrate was concentrated in vacuo. The crude was purified by silica gel chromatography (3% MeOH/DCM) to afford 9-chloro-8-fluoro-N-methyl-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5-amine, Compound 68 (20 mg, 70% yield). ESI-MS m/z: 239.3 [M+H] +.1H NMR (400 MHz, DMSO-d6) δ: 7.47 (br d, J = 2.1 Hz, 1H), 6.93 (br d, J = 10.4 Hz, 1H), 6.41 (br d, J = 2.1 Hz, 1H), 4.32 (br dd, J = 2.7, 12.2 Hz, 1H), 3.89 (br dd, J = 7.0, 12.0 Hz, 1H), 3.13 (br d, J = 13.3 Hz, 2H), 2.82 - 2.75 (m, 1H), 2.36 (s, 3H). Example A8: [0200] Synthesis of (R)-9-chloro-8-fluoro-N,N-dimethyl-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5- amine (Compound 65) and (S)-9-chloro-8-fluoro-N,N-dimethyl-5,6-dihydro-4H-pyrrolo[3,2,1- ij]quinolin-5-amine (Compound 64) [0201] Scheme-7: Synthesis of (R)-9-chloro-8-fluoro-N,N-dimethyl-5,6-dihydro-4H-pyrrolo[3,2,1- ij]quinolin-5-amine (Compound 65) and (S)-9-chloro-8-fluoro-N,N-dimethyl-5,6-dihydro-4H- pyrrolo[3,2,1-ij]quinolin-5-amine (Compound 64)
 [0202] Step-1a: Synthesis of Compound GG-12; tert-butyl (R)-(9-chloro-8-fluoro-5,6-dihydro-4H- pyrrolo[3,2 ,1-ij]quinolin-5-yl)(methyl)carbamate):
[0202] Step-1a: Synthesis of Compound GG-12; tert-butyl (R)-(9-chloro-8-fluoro-5,6-dihydro-4H- pyrrolo[3,2 ,1-ij]quinolin-5-yl)(methyl)carbamate):
 [0205] To a solution of Compound GG-6b-Peak2 (130 mg, 0.40 mmol, 1.0 eq) in DMF (10 vol) at 0 ℃ was added NaH (14.4 mg, 0.60 mmol, 1.5 eq) and the mixture was stirred at 0 ℃ for 10 minutes. MeI (0.049 mL, 0.80 mmol, 2.0 eq) was added at 0 ℃ and the resulting reaction mixture was warmed to room temperature and stirred at that temperature for 2 hours. The reaction mixture was diluted with ice cold water (5 mL) and extracted with ethyl acetate (2 x 5 mL). The combined organic layers were washed with an aqueous solution of NaCl (5 mL), the organic layer was dried over anhydrous Na2SO4, the solids were removed by filtration, and the filtrate was concentrated in vacuo to afford (tert-butyl (S)-(9-chloro-8-fluoro- 5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5-yl)(methyl)carbamate), Compound GG-13 (130 mg, crude) as a pale yellow film. This material was carried forward without further purification. ESI-MS m/z: 338.9 [M+H] +. [0206] Step-2a: Synthesis of (R)-9-chloro-8-fluoro-N,N-dimethyl-5,6-dihydro-4H-pyrrolo[3,2,1- ij]quinolin-5-amine; Compound 65
[0205] To a solution of Compound GG-6b-Peak2 (130 mg, 0.40 mmol, 1.0 eq) in DMF (10 vol) at 0 ℃ was added NaH (14.4 mg, 0.60 mmol, 1.5 eq) and the mixture was stirred at 0 ℃ for 10 minutes. MeI (0.049 mL, 0.80 mmol, 2.0 eq) was added at 0 ℃ and the resulting reaction mixture was warmed to room temperature and stirred at that temperature for 2 hours. The reaction mixture was diluted with ice cold water (5 mL) and extracted with ethyl acetate (2 x 5 mL). The combined organic layers were washed with an aqueous solution of NaCl (5 mL), the organic layer was dried over anhydrous Na2SO4, the solids were removed by filtration, and the filtrate was concentrated in vacuo to afford (tert-butyl (S)-(9-chloro-8-fluoro- 5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5-yl)(methyl)carbamate), Compound GG-13 (130 mg, crude) as a pale yellow film. This material was carried forward without further purification. ESI-MS m/z: 338.9 [M+H] +. [0206] Step-2a: Synthesis of (R)-9-chloro-8-fluoro-N,N-dimethyl-5,6-dihydro-4H-pyrrolo[3,2,1- ij]quinolin-5-amine; Compound 65
 [0207] To a solution of Compound GG-12 (120 mg, 0.35 mmol, 1.0 eq) in DCM (10 vol) at 0 ℃ was added a LiAlH4 solution (2M in THF, 0.71 mL, 1.42 mmol, 4.0 eq). The reaction mixture was heated to reflux and was stirred at that temperature for 2 hours. The reaction mixture was cooled to 0 ℃, quenched with a saturated aqueous NH4Cl solution (10 mL), and extracted with DCM (2 x 10 mL). The combined organic layers were washed with an aqueous solution of NaCl, the organic layer was dried over anhydrous Na2SO4, the solids were filtered, and the filtrate was concentrated in vacuo. The crude material was purified by RP prep HPLC purification to afford (R)-9-chloro-8-fluoro-N,N-dimethyl-5,6-dihydro-4H-pyrrolo[3,2,1-
WSGR Docket No.55776-726.601 ij]quinolin-5-amine, Compound 65 (20 mg, 22% yield). ESI-MS m/z: 253.1 [M+H] +.1H NMR (400 MHz, DMSO-d6) δ: 7.50 - 7.48 (m, 1H), 6.96 (d, J = 10.3 Hz, 1H), 6.42 (d, J = 3.0 Hz, 1H), 4.38 - 4.32 (m, 1H), 4.09 (dd, J = 8.5, 12.3 Hz, 1H), 3.10 - 2.98 (m, 3H), 2.33 - 2.29 (m, 6H). [0208] Step 2b: Synthesis of (S)-9-chloro-8-fluoro-N,N-dimethyl-5,6-dihydro-4H-pyrrolo[3,2,1- ij]quinolin-5-amine (Compound 64)
[0207] To a solution of Compound GG-12 (120 mg, 0.35 mmol, 1.0 eq) in DCM (10 vol) at 0 ℃ was added a LiAlH4 solution (2M in THF, 0.71 mL, 1.42 mmol, 4.0 eq). The reaction mixture was heated to reflux and was stirred at that temperature for 2 hours. The reaction mixture was cooled to 0 ℃, quenched with a saturated aqueous NH4Cl solution (10 mL), and extracted with DCM (2 x 10 mL). The combined organic layers were washed with an aqueous solution of NaCl, the organic layer was dried over anhydrous Na2SO4, the solids were filtered, and the filtrate was concentrated in vacuo. The crude material was purified by RP prep HPLC purification to afford (R)-9-chloro-8-fluoro-N,N-dimethyl-5,6-dihydro-4H-pyrrolo[3,2,1-
WSGR Docket No.55776-726.601 ij]quinolin-5-amine, Compound 65 (20 mg, 22% yield). ESI-MS m/z: 253.1 [M+H] +.1H NMR (400 MHz, DMSO-d6) δ: 7.50 - 7.48 (m, 1H), 6.96 (d, J = 10.3 Hz, 1H), 6.42 (d, J = 3.0 Hz, 1H), 4.38 - 4.32 (m, 1H), 4.09 (dd, J = 8.5, 12.3 Hz, 1H), 3.10 - 2.98 (m, 3H), 2.33 - 2.29 (m, 6H). [0208] Step 2b: Synthesis of (S)-9-chloro-8-fluoro-N,N-dimethyl-5,6-dihydro-4H-pyrrolo[3,2,1- ij]quinolin-5-amine (Compound 64)
 [0212] Step-1: Synthesis of Compound GG-17: tert-butyl (8-chloro-9-fluoro-5,6-dihydro-4H-pyrrolo [3,2,1-ij]quinolin-5-yl)(methyl)carbamate):
WSGR Docket No.55776-726.601
[0212] Step-1: Synthesis of Compound GG-17: tert-butyl (8-chloro-9-fluoro-5,6-dihydro-4H-pyrrolo [3,2,1-ij]quinolin-5-yl)(methyl)carbamate):
WSGR Docket No.55776-726.601
 [0213] To a solution of Compound GG-6d (80 mg, 0.24 mmol, 1.0 eq) in DMF (10 vol) at 0 ℃ was added NaH (8.64 mg, 0.36 mmol, 1.5 eq) and the reaction mixture was stirred at that temperature for 10 minutes. MeI (50.7 mg 0.36 mmol 1.5 eq) was added at 0 ℃ and the resulting mixture was warmed to room temperature and stirred at that temperature for 30 minutes. The reaction mixture was diluted with ice cold water (2 mL) and extracted with ethyl acetate (2 x 2 mL). The combined organic layers were washed with an aqueous solution of NaCl (5 mL), dried over anhydrous Na2SO4, the solids were removed by filtration, and the filtrate was concentrated in vacuo. The crude reaction residue was triturated with diethyl ether (2 x 10 mL), to provide tert-butyl (8-chloro-9-fluoro-5,6-dihydro-4H-pyrrolo [3,2,1-ij]quinolin-5- yl)(methyl)carbamate), Compound GG-17 (90 mg, crude), as a pale brown oil, which was carried forward without further purification. ESI-MS m/z: 338.9 [M+H] +. [0214] Step-2: Synthesis of 8-chloro-9-fluoro-N,N-dimethyl-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin- 5-amine; Compound 70:
[0213] To a solution of Compound GG-6d (80 mg, 0.24 mmol, 1.0 eq) in DMF (10 vol) at 0 ℃ was added NaH (8.64 mg, 0.36 mmol, 1.5 eq) and the reaction mixture was stirred at that temperature for 10 minutes. MeI (50.7 mg 0.36 mmol 1.5 eq) was added at 0 ℃ and the resulting mixture was warmed to room temperature and stirred at that temperature for 30 minutes. The reaction mixture was diluted with ice cold water (2 mL) and extracted with ethyl acetate (2 x 2 mL). The combined organic layers were washed with an aqueous solution of NaCl (5 mL), dried over anhydrous Na2SO4, the solids were removed by filtration, and the filtrate was concentrated in vacuo. The crude reaction residue was triturated with diethyl ether (2 x 10 mL), to provide tert-butyl (8-chloro-9-fluoro-5,6-dihydro-4H-pyrrolo [3,2,1-ij]quinolin-5- yl)(methyl)carbamate), Compound GG-17 (90 mg, crude), as a pale brown oil, which was carried forward without further purification. ESI-MS m/z: 338.9 [M+H] +. [0214] Step-2: Synthesis of 8-chloro-9-fluoro-N,N-dimethyl-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin- 5-amine; Compound 70:
 [0215] To a solution of Compound GG-17 (90 mg, 0.27 mmol, 1.0 eq) in DCM (10 vol) at 0 ℃ was added a LiAlH4 solution (2M in THF, 0.32 mL, 0.65 mmol, 2.5 eq). The resulting reaction mixture was heated to reflux and was stirred at that temperature for 2 hours. The reaction mixture was cooled to 0 ℃ and quenched with a saturated aqueous NH4Cl solution (5 mL). The formed solids were filtered off and washed with multiple portions of ethyl acetate. The combined organic extracts were washed with an aqueous solution of NaCl (10 mL), the organic layer was dried over anhydrous Na2SO4, the solids were filtered, and the filtrate was concentrated in vacuo. The crude was purified by silica gel chromatography (3% MeOH/DCM) to afford 8-chloro-9-fluoro-N,N-dimethyl-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5-amine, Compound 70 (30 mg, 45% yield). ESI-MS m/z: 253.2 [M+H] +.1H NMR (400 MHz, DMSO-d6) δ: 7.40 (s, 1H), 6.97 (d, J = 6.3 Hz, 1H), 6.46 (d, J = 3.0 Hz, 1H), 4.36 - 4.30 (m, 1H), 4.08 (br dd, J = 8.6, 12.2 Hz, 1H), 3.04 - 2.93 (m, 3H), 2.30 (s, 6H).
WSGR Docket No.55776-726.601 Example A10: [0216] Synthesis of 8-chloro-9-fluoro-N-methyl-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5-amine; Compound 69:
[0215] To a solution of Compound GG-17 (90 mg, 0.27 mmol, 1.0 eq) in DCM (10 vol) at 0 ℃ was added a LiAlH4 solution (2M in THF, 0.32 mL, 0.65 mmol, 2.5 eq). The resulting reaction mixture was heated to reflux and was stirred at that temperature for 2 hours. The reaction mixture was cooled to 0 ℃ and quenched with a saturated aqueous NH4Cl solution (5 mL). The formed solids were filtered off and washed with multiple portions of ethyl acetate. The combined organic extracts were washed with an aqueous solution of NaCl (10 mL), the organic layer was dried over anhydrous Na2SO4, the solids were filtered, and the filtrate was concentrated in vacuo. The crude was purified by silica gel chromatography (3% MeOH/DCM) to afford 8-chloro-9-fluoro-N,N-dimethyl-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5-amine, Compound 70 (30 mg, 45% yield). ESI-MS m/z: 253.2 [M+H] +.1H NMR (400 MHz, DMSO-d6) δ: 7.40 (s, 1H), 6.97 (d, J = 6.3 Hz, 1H), 6.46 (d, J = 3.0 Hz, 1H), 4.36 - 4.30 (m, 1H), 4.08 (br dd, J = 8.6, 12.2 Hz, 1H), 3.04 - 2.93 (m, 3H), 2.30 (s, 6H).
WSGR Docket No.55776-726.601 Example A10: [0216] Synthesis of 8-chloro-9-fluoro-N-methyl-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5-amine; Compound 69:
 [0219] To a solution of Compound RP-8 (0.5 g, 1.62 mmol 1.0 eq) in DCM (5 mL) at 0 ℃ was added a LiAlH4 solution (2M in THF, 3.24 mL, 6.49 mmol, 4.0 eq). The reaction mixture was heated to reflux and stirred at that temperature for 2 hours. The reaction mixture was cooled to 0 ℃, quenched with a saturated aqueous NH4Cl solution (5 mL), and extracted with DCM (2 x 10 mL). The combined organic layers were washed with an aqueous solution of NaCl, the organic layer was dried over anhydrous Na2SO4, the solids were filtered, and the filtrate was concentrated in vacuo. The crude material was purified by silica gel
WSGR Docket No.55776-726.601 chromatography (40% EtOAc/Heptane) to afford 8,9-difluoro-N-methyl-5,6-dihydro-4H-pyrrolo[3,2,1- ij]quinolin-5-amine, Compound 72 (280 mg, 77% yield). ESI-MS m/z: 228.2 [M+H] +.1H NMR (400 MHz, DMSO-d6) δ : 7.41 (d, J = 3.0 Hz, 1H), 6.90 (dd, J = 6.9, 11.4 Hz, 1H), 6.47 (d, J = 3.0 Hz, 1H), 4.33 - 4.29 (m, 1H), 3.86 (dd, J = 7.3, 12.1 Hz, 1H), 3.14 - 3.08 (m, 2H), 2.79 - 2.72 (m, 1H), 2.37 (s, 3H). Example A12: [0220] Synthesis of (R)-8,9-difluoro-N-methyl-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5-amine; Compound 63:
[0219] To a solution of Compound RP-8 (0.5 g, 1.62 mmol 1.0 eq) in DCM (5 mL) at 0 ℃ was added a LiAlH4 solution (2M in THF, 3.24 mL, 6.49 mmol, 4.0 eq). The reaction mixture was heated to reflux and stirred at that temperature for 2 hours. The reaction mixture was cooled to 0 ℃, quenched with a saturated aqueous NH4Cl solution (5 mL), and extracted with DCM (2 x 10 mL). The combined organic layers were washed with an aqueous solution of NaCl, the organic layer was dried over anhydrous Na2SO4, the solids were filtered, and the filtrate was concentrated in vacuo. The crude material was purified by silica gel
WSGR Docket No.55776-726.601 chromatography (40% EtOAc/Heptane) to afford 8,9-difluoro-N-methyl-5,6-dihydro-4H-pyrrolo[3,2,1- ij]quinolin-5-amine, Compound 72 (280 mg, 77% yield). ESI-MS m/z: 228.2 [M+H] +.1H NMR (400 MHz, DMSO-d6) δ : 7.41 (d, J = 3.0 Hz, 1H), 6.90 (dd, J = 6.9, 11.4 Hz, 1H), 6.47 (d, J = 3.0 Hz, 1H), 4.33 - 4.29 (m, 1H), 3.86 (dd, J = 7.3, 12.1 Hz, 1H), 3.14 - 3.08 (m, 2H), 2.79 - 2.72 (m, 1H), 2.37 (s, 3H). Example A12: [0220] Synthesis of (R)-8,9-difluoro-N-methyl-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5-amine; Compound 63:
 WSGR Docket No.55776-726.601 [0223] To a solution of Compound RP-10 (250 mg, 0.810 mmol 1.0 eq) in DCM (10 vol) at 0 ℃ was added a LiAlH4 solution (2M in THF, 1.21 mL, 2.43 mmol, 3.0 eq). The resulting reaction mixture was heated to reflux and was stirred at that temperature for 3 hours. The reaction mixture was cooled to 0 ℃ and quenched with a saturated aqueous NH4Cl solution (6 mL). The formed solids were filtered off and washed with multiple portions of ethyl acetate (2 x 6 mL). The combined organic extracts were washed with an aqueous solution of NaCl (10 mL), the organic layer was dried over anhydrous Na2SO4, the solids were filtered, and the filtrate was concentrated in vacuo. The crude was purified by silica gel chromatography (10% MeOH/DCM) to afford (S)-8,9-difluoro-N-methyl-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5- amine, Compound 66 (130 mg, 71% yield). ESI-MS m/z: 223.1 [M+H]+. 1H NMR (400 MHz, METHANOL-d4) δ : 7.24 - 7.22 (m, 1H), 6.83 - 6.77 (m, 1H), 6.47 (d, J = 3.0 Hz, 1H), 4.36 (ddd, J = 0.9 Hz, 3.9 Hz, 12.1 Hz, 1H), 3.97 - 3.92 (m, 1H), 3.29 - 3.20 (m, 2H), 2.85 (dd, J = 7.8 Hz, 15.8 Hz, 1H), 2.49 (s, 3H). Biological Examples Example B-1: Neurite outgrowth assay. [0224] Changes in the pattern of neurite outgrowth have been implicated in neurodegenerative disorders as well as traumatic injuries. The discovery of compounds that can positively affect neuritogenesis are important for developing new therapeutics for neurological diseases. In some instances, measurement of neurite outgrowth of rat cortical neurons using an automated image-based assay is used to determine the neuroplastic effects of the compounds provided herein. In some embodiments, a compound provided herein increases the pattern of neurite outgrowth. In some embodiments, a compound provided herein increases neurite average length compared to a control. In some embodiments, a compound provided herein increases neurite branch points compared to a control. In some embodiments, a compound provided herein increases neurite average length and neurite branch points compared to a control. [0225] In some embodiments, the plastogenic potential of compounds provided herein is assessed by measuring the changes in neurite development. Example B-2: Dendritogenesis Assays. [0226] Phenotypic screening has historically proven more successful than target-based approaches for identifying drugs with novel mechanisms of action. Using a phenotypic assay, the compounds provided herein are tested for their ability to increase dendritic arbor complexity in cultures of cortical neurons. Following treatment, neurons are fixed and visualized using an antibody against MAP2—a cytoskeletal protein localized to the somatodendritic compartment of neurons. Sholl analysis is then performed, and the maximum number of crossings (Nmax) is used as a quantitative metric of dendritic arbor complexity. For statistical comparisons between specific compounds, the raw Nmax values are compared. Percent efficacies are determined by setting the Nmax values for the vehicle (DMSO) and positive (ketamine) controls equal to 0% and 100%, respectively.
WSGR Docket No.55776-726.601 [0227] Animals. For the dendritogenesis experiments, timed pregnant Sprague Dawley rats are obtained from Charles River Laboratories (Wilmington, MA). In some instances, male and female C57BL/6J mice are obtained from Jackson Laboratory (Sacramento, C.A.). In some instances, mice are housed in a temperature and humidity-controlled room maintained on a 12-h light/dark cycle in groups of 4–5 (same sex). Example B-3: Dendritogenesis – Sholl Analysis. [0228] Neurons are plated in 96-well format (200 ^L of media per well) at a density of approximately 15,000 cells/well in Neurobasal (Life Technologies) containing 1% penicillin-streptomycin, 10% heat- inactivated fetal bovine serum, and 0.5 mM glutamine. After 24 h, the medium is replaced with Neurobasal containing 1x B27 supplement (Life Technologies), 1% penicillin-streptomycin, 0.5 mM glutamine, and 12.5 ^M glutamate. After 3 days in vitro (DIV3), the cells are treated with compounds. Compounds tested in the dendritogenesis assays are treated at 10 ^M unless noted otherwise. Stock solutions of the compounds in DMSO are first diluted 100-fold in Neurobasal before an additional 10-fold dilution into each well (total dilution = 1:1000; 0.1% DMSO concentration). Treatments are randomized. After 1 h, the media is removed and replaced with new Neurobasal media containing 1x B27 supplement, 1% penicillin-streptomycin, 0.5 mM glutamine, and 12.5 ^M glutamate. The cells grow for an additional 71 h. At that time, neurons are fixed by removing 80% of the media and replacing it with a volume of 4% aqueous paraformaldehyde (Alfa Aesar) equal to 50% of the working volume of the well. Then, the cells are incubated at room temperature for 20 min before the fixative is aspirated and each well washed twice with DPBS. Cells are permeabilized using 0.2% Triton X-100 (ThermoFisher) in DPBS for 20 minutes at room temperature without shaking. Plates are blocked with antibody diluting buffer (ADB) containing 2% bovine serum albumin (BSA) in DPBS for 1 h at room temperature. Then, plates are incubated overnight at 4ºC with gentle shaking in ADB containing a chicken anti-MAP2 antibody (1:10,000; EnCor, CPCA-MAP2). The next day, plates are washed three times with DPBS and once with 2% ADB in DPBS. Plates are incubated for 1 h at room temperature in ADB containing an anti-chicken IgG secondary antibody conjugated to Alexa Fluor 488 (Life Technologies, 1:500) and washed five times with DPBS. After the final wash, 100 µL of DPBS is added per well and imaged on an ImageXpress Micro XL High-Content Screening System (Molecular Devices, Sunnyvale,CA) with a 20x objective. [0229] Images are analyzed using ImageJ Fiji (version 1.51W). First, images corresponding to each treatment are sorted into individual folders that are then blinded for data analysis. Plate controls (both positive and negative) are used to ensure that the assay is working properly as well as to visually determine appropriate numerical values for brightness/contrast and thresholding to be applied universally to the remainder of the randomized images. Next, the brightness/contrast settings are applied, and approximately 1–2 individual pyramidal-like neurons per image (i.e., no bipolar neurons) are selected using the rectangular selection tool and saved as separate files. Neurons are selected that did not overlap extensively with other cells or extend far beyond the field of view. The threshold settings are then applied to the individual images. The paintbrush tool is used to eliminate artifacts and dendritic processes originating from adjacent neurons
WSGR Docket No.55776-726.601 (cleanup phaseNext, the point tool is used to select the center of the neuron, and the images are saved and processed using the following Sholl analysis batch macro: run("Sholl Analysis...", "starting=0 ending=NaN radius_step=2 #_samples=1 integration=Mean enclosing=1 #_primary=4 infer fit linear polynomial=[Best fitting degree] most semi-log normalizer=Area create background=228 save do"); [0230] Sholl analysis circle radii = 2 pixel increments = 0.67 ^m. All images are taken and analyzed by an experimenter blinded to treatment conditions. The number of crossings for each neuron at each distinct radius is averaged to produce an average Sholl plot for each treatment. The Nmax values are simply
WSGR Docket No.55776-726.601 [0223] To a solution of Compound RP-10 (250 mg, 0.810 mmol 1.0 eq) in DCM (10 vol) at 0 ℃ was added a LiAlH4 solution (2M in THF, 1.21 mL, 2.43 mmol, 3.0 eq). The resulting reaction mixture was heated to reflux and was stirred at that temperature for 3 hours. The reaction mixture was cooled to 0 ℃ and quenched with a saturated aqueous NH4Cl solution (6 mL). The formed solids were filtered off and washed with multiple portions of ethyl acetate (2 x 6 mL). The combined organic extracts were washed with an aqueous solution of NaCl (10 mL), the organic layer was dried over anhydrous Na2SO4, the solids were filtered, and the filtrate was concentrated in vacuo. The crude was purified by silica gel chromatography (10% MeOH/DCM) to afford (S)-8,9-difluoro-N-methyl-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-5- amine, Compound 66 (130 mg, 71% yield). ESI-MS m/z: 223.1 [M+H]+. 1H NMR (400 MHz, METHANOL-d4) δ : 7.24 - 7.22 (m, 1H), 6.83 - 6.77 (m, 1H), 6.47 (d, J = 3.0 Hz, 1H), 4.36 (ddd, J = 0.9 Hz, 3.9 Hz, 12.1 Hz, 1H), 3.97 - 3.92 (m, 1H), 3.29 - 3.20 (m, 2H), 2.85 (dd, J = 7.8 Hz, 15.8 Hz, 1H), 2.49 (s, 3H). Biological Examples Example B-1: Neurite outgrowth assay. [0224] Changes in the pattern of neurite outgrowth have been implicated in neurodegenerative disorders as well as traumatic injuries. The discovery of compounds that can positively affect neuritogenesis are important for developing new therapeutics for neurological diseases. In some instances, measurement of neurite outgrowth of rat cortical neurons using an automated image-based assay is used to determine the neuroplastic effects of the compounds provided herein. In some embodiments, a compound provided herein increases the pattern of neurite outgrowth. In some embodiments, a compound provided herein increases neurite average length compared to a control. In some embodiments, a compound provided herein increases neurite branch points compared to a control. In some embodiments, a compound provided herein increases neurite average length and neurite branch points compared to a control. [0225] In some embodiments, the plastogenic potential of compounds provided herein is assessed by measuring the changes in neurite development. Example B-2: Dendritogenesis Assays. [0226] Phenotypic screening has historically proven more successful than target-based approaches for identifying drugs with novel mechanisms of action. Using a phenotypic assay, the compounds provided herein are tested for their ability to increase dendritic arbor complexity in cultures of cortical neurons. Following treatment, neurons are fixed and visualized using an antibody against MAP2—a cytoskeletal protein localized to the somatodendritic compartment of neurons. Sholl analysis is then performed, and the maximum number of crossings (Nmax) is used as a quantitative metric of dendritic arbor complexity. For statistical comparisons between specific compounds, the raw Nmax values are compared. Percent efficacies are determined by setting the Nmax values for the vehicle (DMSO) and positive (ketamine) controls equal to 0% and 100%, respectively.
WSGR Docket No.55776-726.601 [0227] Animals. For the dendritogenesis experiments, timed pregnant Sprague Dawley rats are obtained from Charles River Laboratories (Wilmington, MA). In some instances, male and female C57BL/6J mice are obtained from Jackson Laboratory (Sacramento, C.A.). In some instances, mice are housed in a temperature and humidity-controlled room maintained on a 12-h light/dark cycle in groups of 4–5 (same sex). Example B-3: Dendritogenesis – Sholl Analysis. [0228] Neurons are plated in 96-well format (200 ^L of media per well) at a density of approximately 15,000 cells/well in Neurobasal (Life Technologies) containing 1% penicillin-streptomycin, 10% heat- inactivated fetal bovine serum, and 0.5 mM glutamine. After 24 h, the medium is replaced with Neurobasal containing 1x B27 supplement (Life Technologies), 1% penicillin-streptomycin, 0.5 mM glutamine, and 12.5 ^M glutamate. After 3 days in vitro (DIV3), the cells are treated with compounds. Compounds tested in the dendritogenesis assays are treated at 10 ^M unless noted otherwise. Stock solutions of the compounds in DMSO are first diluted 100-fold in Neurobasal before an additional 10-fold dilution into each well (total dilution = 1:1000; 0.1% DMSO concentration). Treatments are randomized. After 1 h, the media is removed and replaced with new Neurobasal media containing 1x B27 supplement, 1% penicillin-streptomycin, 0.5 mM glutamine, and 12.5 ^M glutamate. The cells grow for an additional 71 h. At that time, neurons are fixed by removing 80% of the media and replacing it with a volume of 4% aqueous paraformaldehyde (Alfa Aesar) equal to 50% of the working volume of the well. Then, the cells are incubated at room temperature for 20 min before the fixative is aspirated and each well washed twice with DPBS. Cells are permeabilized using 0.2% Triton X-100 (ThermoFisher) in DPBS for 20 minutes at room temperature without shaking. Plates are blocked with antibody diluting buffer (ADB) containing 2% bovine serum albumin (BSA) in DPBS for 1 h at room temperature. Then, plates are incubated overnight at 4ºC with gentle shaking in ADB containing a chicken anti-MAP2 antibody (1:10,000; EnCor, CPCA-MAP2). The next day, plates are washed three times with DPBS and once with 2% ADB in DPBS. Plates are incubated for 1 h at room temperature in ADB containing an anti-chicken IgG secondary antibody conjugated to Alexa Fluor 488 (Life Technologies, 1:500) and washed five times with DPBS. After the final wash, 100 µL of DPBS is added per well and imaged on an ImageXpress Micro XL High-Content Screening System (Molecular Devices, Sunnyvale,CA) with a 20x objective. [0229] Images are analyzed using ImageJ Fiji (version 1.51W). First, images corresponding to each treatment are sorted into individual folders that are then blinded for data analysis. Plate controls (both positive and negative) are used to ensure that the assay is working properly as well as to visually determine appropriate numerical values for brightness/contrast and thresholding to be applied universally to the remainder of the randomized images. Next, the brightness/contrast settings are applied, and approximately 1–2 individual pyramidal-like neurons per image (i.e., no bipolar neurons) are selected using the rectangular selection tool and saved as separate files. Neurons are selected that did not overlap extensively with other cells or extend far beyond the field of view. The threshold settings are then applied to the individual images. The paintbrush tool is used to eliminate artifacts and dendritic processes originating from adjacent neurons
WSGR Docket No.55776-726.601 (cleanup phaseNext, the point tool is used to select the center of the neuron, and the images are saved and processed using the following Sholl analysis batch macro: run("Sholl Analysis...", "starting=0 ending=NaN radius_step=2 #_samples=1 integration=Mean enclosing=1 #_primary=4 infer fit linear polynomial=[Best fitting degree] most semi-log normalizer=Area create background=228 save do"); [0230] Sholl analysis circle radii = 2 pixel increments = 0.67 ^m. All images are taken and analyzed by an experimenter blinded to treatment conditions. The number of crossings for each neuron at each distinct radius is averaged to produce an average Sholl plot for each treatment. The Nmax values are simply
 Formula (I) wherein: each R3, R4, and R5 is independently hydrogen, C1-C6alkyl, C1-C6haloalkyl, C1-C6hydroxyalkyl, C1- C6aminoalkyl, C1-C6heteroalkyl, halogen, -CN, -NO2, -ORa, -SRa, -NRcRd, -S(=O)Rb, -S(=O)2Rb, - S
Formula (I) wherein: each R3, R4, and R5 is independently hydrogen, C1-C6alkyl, C1-C6haloalkyl, C1-C6hydroxyalkyl, C1- C6aminoalkyl, C1-C6heteroalkyl, halogen, -CN, -NO2, -ORa, -SRa, -NRcRd, -S(=O)Rb, -S(=O)2Rb, - S
 - P(=O)(ORc)(ORd), -P(=O)RcRd, aryl, heteroaryl, cycloalkyl, heterocycloalkyl, C1-C6alkyl(cycloalkyl), C1-C6alkyl(heterocycloalkyl), C1-C6alkyl(aryl), or C1-C6alkyl(heteroaryl), wherein each of the alkyl, heteroalkyl, aryl, heteroaryl, cycloalkyl, and heterocycloalkyl is optionally substituted with one or more substituents selected from C1-C3alkyl, C1-C3haloalkyl, C1- C3alkoxy, halogen, -OH, -CN, and =O; R8 is hydrogen or C1-C6alkyl; R9 is hydrogen or C1-C6alkyl; or R8 and R9 are taken together to form a heterocycloalkyl, wherein the heterocycloalkyl is optionally substituted with one or more substituents selected from C1-C3alkyl, C1-C3haloalkyl, C1-C3alkoxy, halogen, -OH, and =O; each Ra is independently hydrogen, C1-C6alkyl, C1-C6haloalkyl, C1-C6heteroalkyl, aryl, C1- C3alkyl(phenyl), C3-C6cycloalkyl, 5- to 6- membered heteroaryl, C1-C3alkyl(5- to 6- membered heteroaryl), or 4- to 6- membered heterocycloalkyl, wherein each alkyl, heteroalkyl, aryl, cycloalkyl, heteroaryl, and heterocycloalkyl is optionally substituted with one or more substituents selected from C1-C3alkyl, C1-C3haloalkyl, C1-C3alkoxy, halogen, -OH, -CN, and =O; each Rb is independently hydrogen or C1-C6alkyl; and each Rc and Rd is independently hydrogen or C1-C6alkyl; or
WSGR Docket No.55776-726.601 Rc and Rd are taken together to form a 4- to 6- membered heterocycloalkyl, wherein the heterocycloalkyl is optionally substituted with one or more substituents selected from C1-C3alkyl, C1-C3haloalkyl, C1- C3alkoxy, halogen, -OH, -CN, and =O; whereinat least two of R3, R4, and R5 are not hydrogen. 2. The compound of claim 1, or a pharmaceutically acceptable salt, pharmaceutically acceptable solvate, diastereomeric mixture, individual enantiomer, or isotopically enriched form thereof, wherein: each R3, R4, and R5 is independently hydrogen, C1-C6alkyl, C1-C6haloalkyl, C1-C6hydroxyalkyl, C1- C6aminoalkyl, C1-C6heteroalkyl, halogen, -CN, -NO2, -ORa, -SRa, -NRcRd, -S(=O)Rb, -S(=O)2Rb, - S(=O)2NRcRd, -NRbS(=O)2Rb, -NRbS(=O)2NRcRd, -C(=O)Rb, -C(=O)ORb, -OC(=O)Rb, - OC(=O)ORb, -OC(=O)NRcRd, -NRbC(=O)Rb, -NRbC(=O)ORb, -NRbC(=O)NRcRd, -C(=O)NRcRd, - P(=O)(ORc)(ORd), -P(=O)RcRd, C6-C10aryl, 5-10 membered heteroaryl, C3-C7cycloalkyl, 3- to 10- membered heterocycloalkyl, C1-C6alkyl(C3-C7cycloalkyl), C1-C6alkyl(3- to 10- membered heterocycloalkyl), C1-C6alkyl(C6-C10aryl), or C1-C6alkyl(heteroaryl), wherein each of the alkyl, heteroalkyl, aryl, heteroaryl, cycloalkyl, and heterocycloalkyl is optionally substituted with one or more substituents selected from C1-C3alkyl, C1-C3haloalkyl, C1-C3alkoxy, halogen, -OH, -CN, and =O. 3 The compound of claim 1 or 2, or a pharmaceutically acceptable salt, pharmaceutically acceptable solvate, diastereomeric mixture, individual enantiomer, or isotopically enriched form thereof, wherein: each R3, R4, and R5 is independently hydrogen, C1-C6alkyl, C1-C6heteroalkyl, halogen, C6-C10aryl, or C3- C7cycloalkyl, wherein each of the alkyl, heteroalkyl, aryl, and cycloalkyl is optionally substituted with one or more substituents selected from halogen, C1-C3alkyl, C1-C3alkoxy, halogen, -OH, and - CN. 4 The compound of any one of claims 1 to 3, or a pharmaceutically acceptable salt, pharmaceutically acceptable solvate, diastereomeric mixture, individual enantiomer, or isotopically enriched form thereof, wherein: each R3, R4, and R5 is independently hydrogen, C1-C6alkyl, C1-C6heteroalkyl, -F, -Cl, -Br, phenyl, cyclopropyl, or cyclobutyl, wherein each of the alkyl, heteroalkyl, phenyl, cyclopropyl, and cyclobutyl is optionally substituted with one or more substituents selected from C1-C3alkyl, C1- C3alkoxy, -F, -Cl, -Br, and -CN. 5 The compound of any one of claims 1 to 4, or a pharmaceutically acceptable salt, pharmaceutically acceptable solvate, diastereomeric mixture, individual enantiomer, or isotopically enriched form thereof, wherein: R3 is hydrogen, C1-C6alkyl, C1-C6heteroalkyl, -F, -Cl, -Br, or phenyl, wherein each of the alkyl, heteroalkyl, and phenyl is optionally substituted with one or more substituents selected from C1- C3alkyl, C1-C3alkoxy, -F, -Cl, -Br, and -CN.
WSGR Docket No.55776-726.601 6. The compound of any one of claims 1 to 5, or a pharmaceutically acceptable salt, pharmaceutically acceptable solvate, diastereomeric mixture, individual enantiomer, or isotopically enriched form thereof, wherein: R3 is hydrogen. 7. The compound of any one of claims 1 to 5, or a pharmaceutically acceptable salt, pharmaceutically acceptable solvate, diastereomeric mixture, individual enantiomer, or isotopically enriched form thereof, wherein: R3 is C1-C6alkyl, C1-C6heteroalkyl, -F, -Cl, -Br, or phenyl, wherein each of the alkyl, heteroalkyl, and phenyl is optionally substituted with one or more substituents selected from C1-C3alkyl, C1- C3alkoxy, -F, -Cl, -Br, and -CN. 8 The compound of any one of claims 1 to 7, or a pharmaceutically acceptable salt, pharmaceutically acceptable solvate, diastereomeric mixture, individual enantiomer, or isotopically enriched form thereof, wherein: R4 is hydrogen, C1-C6alkyl, C1-C6heteroalkyl, -F, -Cl, -Br, or phenyl, wherein each of the alkyl, heteroalkyl, and phenyl is optionally substituted with one or more substituents selected from C1- C3alkyl, C1-C3alkoxy, -F, -Cl, -Br, and -CN. 9 The compound of any one of claims 1 to 8, or a pharmaceutically acceptable salt, pharmaceutically acceptable solvate, diastereomeric mixture, individual enantiomer, or isotopically enriched form thereof, wherein: R4 is hydrogen. 10 The compound of any one of claims 1 to 8, or a pharmaceutically acceptable salt, pharmaceutically acceptable solvate, diastereomeric mixture, individual enantiomer, or isotopically enriched form thereof, wherein: R4 is C1-C6alkyl, C1-C6heteroalkyl, -F, -Cl, -Br, or phenyl, wherein each of the alkyl, heteroalkyl, and phenyl is optionally substituted with one or more substituents selected from C1-C3alkyl, C1- C3alkoxy, -F, -Cl, -Br, and -CN. 11 The compound of any one of claims 1 to 10, or a pharmaceutically acceptable salt, pharmaceutically acceptable solvate, diastereomeric mixture, individual enantiomer, or isotopically enriched form thereof, wherein: R5 is hydrogen, C1-C6alkyl, C1-C6heteroalkyl, -F, -Cl, -Br, or phenyl, wherein each of the alkyl, heteroalkyl, and phenyl is optionally substituted with one or more substituents selected from C1- C3alkyl, C1-C3alkoxy, -F, -Cl, -Br, and -CN. 12 The compound of any one of claims 1 to 11, or a pharmaceutically acceptable salt, pharmaceutically acceptable solvate, diastereomeric mixture, individual enantiomer, or isotopically enriched form thereof, wherein: R5 is hydrogen.
WSGR Docket No.55776-726.601 13. The compound of any one of claims 1 to 11, or a pharmaceutically acceptable salt, pharmaceutically acceptable solvate, diastereomeric mixture, individual enantiomer, or isotopically enriched form thereof, wherein: R5 is C1-C6alkyl, C1-C6heteroalkyl, -F, -Cl, -Br, or phenyl, wherein each of the alkyl, heteroalkyl, and phenyl is optionally substituted with one or more substituents selected from C1-C3alkyl, C1- C3alkoxy, -F, -Cl, -Br, and -CN. 14. The compound of any one of claims 1 to 4, or a pharmaceutically acceptable salt, pharmaceutically acceptable solvate, diastereomeric mixture, individual enantiomer, or isotopically enriched form thereof, wherein: each R3, R4, and R5 is independently hydrogen, -CH3, -F, -Cl, or -Br. 15. The compound of any one of claims 1 to 14, or a pharmaceutically acceptable salt, pharmaceutically acceptable solvate, diastereomeric mixture, individual enantiomer, or isotopically enriched form thereof, wherein: R8 is hydrogen or C1-C3alkyl. 16. The compound of any one of claims 1 to 15, or a pharmaceutically acceptable salt, pharmaceutically acceptable solvate, diastereomeric mixture, individual enantiomer, or isotopically enriched form thereof, wherein: R8 is hydrogen. 17. The compound of any one of claims 1 to 15, or a pharmaceutically acceptable salt, pharmaceutically acceptable solvate, diastereomeric mixture, individual enantiomer, or isotopically enriched form thereof, wherein: R8 is C1-C3alkyl. 18. The compound of any one of claims 1 to 17, or a pharmaceutically acceptable salt, pharmaceutically acceptable solvate, diastereomeric mixture, individual enantiomer, or isotopically enriched form thereof, wherein: R9 is hydrogen or C1-C3alkyl. 19. The compound of any one of claims 1 to 18, or a pharmaceutically acceptable salt, pharmaceutically acceptable solvate, diastereomeric mixture, individual enantiomer, or isotopically enriched form thereof, wherein: R9 is hydrogen. 20. The compound of any one of claims 1 to 18, or a pharmaceutically acceptable salt, pharmaceutically acceptable solvate, diastereomeric mixture, individual enantiomer, or isotopically enriched form thereof, wherein: R9 is -CH3. 21. The compound of any one of claims 1 to 20, or a pharmaceutically acceptable salt, pharmaceutically acceptable solvate, diastereomeric mixture, individual enantiomer, or isotopically enriched form thereof, wherein:
WSGR Docket No.55776-726.601 R9 is -CH2CH3. 22. A compound of claim, or a pharmaceutically acceptable salt, pharmaceutically acceptable solvate, diastereomeric mixture, individual enantiomer, or isotopically enriched form thereof, wherein the compound is: or
- P(=O)(ORc)(ORd), -P(=O)RcRd, aryl, heteroaryl, cycloalkyl, heterocycloalkyl, C1-C6alkyl(cycloalkyl), C1-C6alkyl(heterocycloalkyl), C1-C6alkyl(aryl), or C1-C6alkyl(heteroaryl), wherein each of the alkyl, heteroalkyl, aryl, heteroaryl, cycloalkyl, and heterocycloalkyl is optionally substituted with one or more substituents selected from C1-C3alkyl, C1-C3haloalkyl, C1- C3alkoxy, halogen, -OH, -CN, and =O; R8 is hydrogen or C1-C6alkyl; R9 is hydrogen or C1-C6alkyl; or R8 and R9 are taken together to form a heterocycloalkyl, wherein the heterocycloalkyl is optionally substituted with one or more substituents selected from C1-C3alkyl, C1-C3haloalkyl, C1-C3alkoxy, halogen, -OH, and =O; each Ra is independently hydrogen, C1-C6alkyl, C1-C6haloalkyl, C1-C6heteroalkyl, aryl, C1- C3alkyl(phenyl), C3-C6cycloalkyl, 5- to 6- membered heteroaryl, C1-C3alkyl(5- to 6- membered heteroaryl), or 4- to 6- membered heterocycloalkyl, wherein each alkyl, heteroalkyl, aryl, cycloalkyl, heteroaryl, and heterocycloalkyl is optionally substituted with one or more substituents selected from C1-C3alkyl, C1-C3haloalkyl, C1-C3alkoxy, halogen, -OH, -CN, and =O; each Rb is independently hydrogen or C1-C6alkyl; and each Rc and Rd is independently hydrogen or C1-C6alkyl; or
WSGR Docket No.55776-726.601 Rc and Rd are taken together to form a 4- to 6- membered heterocycloalkyl, wherein the heterocycloalkyl is optionally substituted with one or more substituents selected from C1-C3alkyl, C1-C3haloalkyl, C1- C3alkoxy, halogen, -OH, -CN, and =O; whereinat least two of R3, R4, and R5 are not hydrogen. 2. The compound of claim 1, or a pharmaceutically acceptable salt, pharmaceutically acceptable solvate, diastereomeric mixture, individual enantiomer, or isotopically enriched form thereof, wherein: each R3, R4, and R5 is independently hydrogen, C1-C6alkyl, C1-C6haloalkyl, C1-C6hydroxyalkyl, C1- C6aminoalkyl, C1-C6heteroalkyl, halogen, -CN, -NO2, -ORa, -SRa, -NRcRd, -S(=O)Rb, -S(=O)2Rb, - S(=O)2NRcRd, -NRbS(=O)2Rb, -NRbS(=O)2NRcRd, -C(=O)Rb, -C(=O)ORb, -OC(=O)Rb, - OC(=O)ORb, -OC(=O)NRcRd, -NRbC(=O)Rb, -NRbC(=O)ORb, -NRbC(=O)NRcRd, -C(=O)NRcRd, - P(=O)(ORc)(ORd), -P(=O)RcRd, C6-C10aryl, 5-10 membered heteroaryl, C3-C7cycloalkyl, 3- to 10- membered heterocycloalkyl, C1-C6alkyl(C3-C7cycloalkyl), C1-C6alkyl(3- to 10- membered heterocycloalkyl), C1-C6alkyl(C6-C10aryl), or C1-C6alkyl(heteroaryl), wherein each of the alkyl, heteroalkyl, aryl, heteroaryl, cycloalkyl, and heterocycloalkyl is optionally substituted with one or more substituents selected from C1-C3alkyl, C1-C3haloalkyl, C1-C3alkoxy, halogen, -OH, -CN, and =O. 3 The compound of claim 1 or 2, or a pharmaceutically acceptable salt, pharmaceutically acceptable solvate, diastereomeric mixture, individual enantiomer, or isotopically enriched form thereof, wherein: each R3, R4, and R5 is independently hydrogen, C1-C6alkyl, C1-C6heteroalkyl, halogen, C6-C10aryl, or C3- C7cycloalkyl, wherein each of the alkyl, heteroalkyl, aryl, and cycloalkyl is optionally substituted with one or more substituents selected from halogen, C1-C3alkyl, C1-C3alkoxy, halogen, -OH, and - CN. 4 The compound of any one of claims 1 to 3, or a pharmaceutically acceptable salt, pharmaceutically acceptable solvate, diastereomeric mixture, individual enantiomer, or isotopically enriched form thereof, wherein: each R3, R4, and R5 is independently hydrogen, C1-C6alkyl, C1-C6heteroalkyl, -F, -Cl, -Br, phenyl, cyclopropyl, or cyclobutyl, wherein each of the alkyl, heteroalkyl, phenyl, cyclopropyl, and cyclobutyl is optionally substituted with one or more substituents selected from C1-C3alkyl, C1- C3alkoxy, -F, -Cl, -Br, and -CN. 5 The compound of any one of claims 1 to 4, or a pharmaceutically acceptable salt, pharmaceutically acceptable solvate, diastereomeric mixture, individual enantiomer, or isotopically enriched form thereof, wherein: R3 is hydrogen, C1-C6alkyl, C1-C6heteroalkyl, -F, -Cl, -Br, or phenyl, wherein each of the alkyl, heteroalkyl, and phenyl is optionally substituted with one or more substituents selected from C1- C3alkyl, C1-C3alkoxy, -F, -Cl, -Br, and -CN.
WSGR Docket No.55776-726.601 6. The compound of any one of claims 1 to 5, or a pharmaceutically acceptable salt, pharmaceutically acceptable solvate, diastereomeric mixture, individual enantiomer, or isotopically enriched form thereof, wherein: R3 is hydrogen. 7. The compound of any one of claims 1 to 5, or a pharmaceutically acceptable salt, pharmaceutically acceptable solvate, diastereomeric mixture, individual enantiomer, or isotopically enriched form thereof, wherein: R3 is C1-C6alkyl, C1-C6heteroalkyl, -F, -Cl, -Br, or phenyl, wherein each of the alkyl, heteroalkyl, and phenyl is optionally substituted with one or more substituents selected from C1-C3alkyl, C1- C3alkoxy, -F, -Cl, -Br, and -CN. 8 The compound of any one of claims 1 to 7, or a pharmaceutically acceptable salt, pharmaceutically acceptable solvate, diastereomeric mixture, individual enantiomer, or isotopically enriched form thereof, wherein: R4 is hydrogen, C1-C6alkyl, C1-C6heteroalkyl, -F, -Cl, -Br, or phenyl, wherein each of the alkyl, heteroalkyl, and phenyl is optionally substituted with one or more substituents selected from C1- C3alkyl, C1-C3alkoxy, -F, -Cl, -Br, and -CN. 9 The compound of any one of claims 1 to 8, or a pharmaceutically acceptable salt, pharmaceutically acceptable solvate, diastereomeric mixture, individual enantiomer, or isotopically enriched form thereof, wherein: R4 is hydrogen. 10 The compound of any one of claims 1 to 8, or a pharmaceutically acceptable salt, pharmaceutically acceptable solvate, diastereomeric mixture, individual enantiomer, or isotopically enriched form thereof, wherein: R4 is C1-C6alkyl, C1-C6heteroalkyl, -F, -Cl, -Br, or phenyl, wherein each of the alkyl, heteroalkyl, and phenyl is optionally substituted with one or more substituents selected from C1-C3alkyl, C1- C3alkoxy, -F, -Cl, -Br, and -CN. 11 The compound of any one of claims 1 to 10, or a pharmaceutically acceptable salt, pharmaceutically acceptable solvate, diastereomeric mixture, individual enantiomer, or isotopically enriched form thereof, wherein: R5 is hydrogen, C1-C6alkyl, C1-C6heteroalkyl, -F, -Cl, -Br, or phenyl, wherein each of the alkyl, heteroalkyl, and phenyl is optionally substituted with one or more substituents selected from C1- C3alkyl, C1-C3alkoxy, -F, -Cl, -Br, and -CN. 12 The compound of any one of claims 1 to 11, or a pharmaceutically acceptable salt, pharmaceutically acceptable solvate, diastereomeric mixture, individual enantiomer, or isotopically enriched form thereof, wherein: R5 is hydrogen.
WSGR Docket No.55776-726.601 13. The compound of any one of claims 1 to 11, or a pharmaceutically acceptable salt, pharmaceutically acceptable solvate, diastereomeric mixture, individual enantiomer, or isotopically enriched form thereof, wherein: R5 is C1-C6alkyl, C1-C6heteroalkyl, -F, -Cl, -Br, or phenyl, wherein each of the alkyl, heteroalkyl, and phenyl is optionally substituted with one or more substituents selected from C1-C3alkyl, C1- C3alkoxy, -F, -Cl, -Br, and -CN. 14. The compound of any one of claims 1 to 4, or a pharmaceutically acceptable salt, pharmaceutically acceptable solvate, diastereomeric mixture, individual enantiomer, or isotopically enriched form thereof, wherein: each R3, R4, and R5 is independently hydrogen, -CH3, -F, -Cl, or -Br. 15. The compound of any one of claims 1 to 14, or a pharmaceutically acceptable salt, pharmaceutically acceptable solvate, diastereomeric mixture, individual enantiomer, or isotopically enriched form thereof, wherein: R8 is hydrogen or C1-C3alkyl. 16. The compound of any one of claims 1 to 15, or a pharmaceutically acceptable salt, pharmaceutically acceptable solvate, diastereomeric mixture, individual enantiomer, or isotopically enriched form thereof, wherein: R8 is hydrogen. 17. The compound of any one of claims 1 to 15, or a pharmaceutically acceptable salt, pharmaceutically acceptable solvate, diastereomeric mixture, individual enantiomer, or isotopically enriched form thereof, wherein: R8 is C1-C3alkyl. 18. The compound of any one of claims 1 to 17, or a pharmaceutically acceptable salt, pharmaceutically acceptable solvate, diastereomeric mixture, individual enantiomer, or isotopically enriched form thereof, wherein: R9 is hydrogen or C1-C3alkyl. 19. The compound of any one of claims 1 to 18, or a pharmaceutically acceptable salt, pharmaceutically acceptable solvate, diastereomeric mixture, individual enantiomer, or isotopically enriched form thereof, wherein: R9 is hydrogen. 20. The compound of any one of claims 1 to 18, or a pharmaceutically acceptable salt, pharmaceutically acceptable solvate, diastereomeric mixture, individual enantiomer, or isotopically enriched form thereof, wherein: R9 is -CH3. 21. The compound of any one of claims 1 to 20, or a pharmaceutically acceptable salt, pharmaceutically acceptable solvate, diastereomeric mixture, individual enantiomer, or isotopically enriched form thereof, wherein:
WSGR Docket No.55776-726.601 R9 is -CH2CH3. 22. A compound of claim, or a pharmaceutically acceptable salt, pharmaceutically acceptable solvate, diastereomeric mixture, individual enantiomer, or isotopically enriched form thereof, wherein the compound is: or
 . 23. The compound of any one of claims 1 to 22 or a pharmaceutically acceptable salt, pharmaceutically acceptable solvate, diastereomeric mixture, individual enantiomer, or isotopically enriched form thereof, for use as medicine. 24. A pharmaceutical composition comprising the compound of any one of claims 1 to 22, or a pharmaceutically acceptable salt, pharmaceutically acceptable solvate, diastereomeric mixture, individual enantiomer, or isotopically enriched form thereof. 25. The pharmaceutical composition of claim 24, further comprising at least one pharmaceutically acceptable excipient. 26. A method of treating a disease or disorder in a subject in need thereof, the method comprising administering to the subject a therapeutically effective amount of the compound of any one of claims 1- 22, or a pharmaceutically acceptable salt, pharmaceutically acceptable solvate, diastereomeric mixture, individual enantiomer, or isotopically enriched form thereof.
WSGR Docket No.55776-726.601 27. The method of claim 26, wherein the disease or disorder is a disease or disorder of the brain. 28. The method of claim 26, wherein the disease or disorder is a neurodegenerative, a neuropsychiatric, or substance use disease or disorder. 29. The method of claim 26, wherein the disease or disorder is a neurological disease or disorder. 30. The method of claim 26, wherein the disease or disorder is a brain injury. 31. The method of claim 26 or 27, wherein the disease or disorder is an anxiety disorder, a mood disorder, a psychotic disorder, a personality disorder, an eating disorder, a sleep disorder, a sexuality disorder, an impulse control disorder, a substance use disorder, a dissociative disorder, a cognitive disorder, a developmental disorder, or a factitious disorder. 32. The method of claim 26 or 27, wherein the disease or disorder is a psychotic disorder. 33. The method of claim 31 or 32, wherein the psychotic disorder is selected from schizophrenia, schizoaffective disorder, schizophreniform disorder, brief psychotic disorder, delusional disorder, shared psychotic disorder, substance-induced psychotic disorder, paraphrenia, psychotic depression, bipolar disorder, schizotypal personality disorder, paranoid personality disorder, schizoid personality disorder, borderline personality disorder, post-traumatic stress disorder, obsessive-compulsive disorder, and dissociative disorders, or psychosis associated with a neurodegenerative disorders. 34. The method of claim 33, wherein the neurodegenerative disorder is selected from Huntington’s disease, Alzheimer’s disease, Lewy body dementia, and Parkinson’s disease. 35. The method of claim 32 or 33, wherein the psychotic disorder is schizophrenia or bipolar disorder. 36. The method of any one of claims 26 to 35, wherein the method further comprises administering to the subject a therapeutically effective amount of an additional therapeutic agent.
. 23. The compound of any one of claims 1 to 22 or a pharmaceutically acceptable salt, pharmaceutically acceptable solvate, diastereomeric mixture, individual enantiomer, or isotopically enriched form thereof, for use as medicine. 24. A pharmaceutical composition comprising the compound of any one of claims 1 to 22, or a pharmaceutically acceptable salt, pharmaceutically acceptable solvate, diastereomeric mixture, individual enantiomer, or isotopically enriched form thereof. 25. The pharmaceutical composition of claim 24, further comprising at least one pharmaceutically acceptable excipient. 26. A method of treating a disease or disorder in a subject in need thereof, the method comprising administering to the subject a therapeutically effective amount of the compound of any one of claims 1- 22, or a pharmaceutically acceptable salt, pharmaceutically acceptable solvate, diastereomeric mixture, individual enantiomer, or isotopically enriched form thereof.
WSGR Docket No.55776-726.601 27. The method of claim 26, wherein the disease or disorder is a disease or disorder of the brain. 28. The method of claim 26, wherein the disease or disorder is a neurodegenerative, a neuropsychiatric, or substance use disease or disorder. 29. The method of claim 26, wherein the disease or disorder is a neurological disease or disorder. 30. The method of claim 26, wherein the disease or disorder is a brain injury. 31. The method of claim 26 or 27, wherein the disease or disorder is an anxiety disorder, a mood disorder, a psychotic disorder, a personality disorder, an eating disorder, a sleep disorder, a sexuality disorder, an impulse control disorder, a substance use disorder, a dissociative disorder, a cognitive disorder, a developmental disorder, or a factitious disorder. 32. The method of claim 26 or 27, wherein the disease or disorder is a psychotic disorder. 33. The method of claim 31 or 32, wherein the psychotic disorder is selected from schizophrenia, schizoaffective disorder, schizophreniform disorder, brief psychotic disorder, delusional disorder, shared psychotic disorder, substance-induced psychotic disorder, paraphrenia, psychotic depression, bipolar disorder, schizotypal personality disorder, paranoid personality disorder, schizoid personality disorder, borderline personality disorder, post-traumatic stress disorder, obsessive-compulsive disorder, and dissociative disorders, or psychosis associated with a neurodegenerative disorders. 34. The method of claim 33, wherein the neurodegenerative disorder is selected from Huntington’s disease, Alzheimer’s disease, Lewy body dementia, and Parkinson’s disease. 35. The method of claim 32 or 33, wherein the psychotic disorder is schizophrenia or bipolar disorder. 36. The method of any one of claims 26 to 35, wherein the method further comprises administering to the subject a therapeutically effective amount of an additional therapeutic agent.